
蛋白质折叠速率决定因素与预测方法的研究进展
2013-09-20 09:08徐宏睿马彬广
Biophysics Reports 2013年3期
徐宏睿, 马彬广
华中农业大学生命科学技术学院生物信息中心,武汉 43007
引 言
蛋白质是由氨基酸按照一定的顺序组成的生物大分子,在生物体内担当着重要的角色。自然状态下,蛋白质通常能从未折叠状态快速而可靠地折叠成具有三维结构的天然构象。正确的结构是功能的基础,蛋白质的错误折叠会形成无活性蛋白或引起淀粉样纤维的聚集,引起阿尔茨海默病、帕金森病、亨廷顿病、传染性海绵状脑病等蛋白质折叠疾病[1,2]。因而,深入了解和研究蛋白质折叠问题,成为近年来科学家们不懈努力的方向。
因为组成蛋白质的20种氨基酸残基的异质性及蛋白质结构的多样性,蛋白质折叠是一个极其复杂的过程[3],涉及氢键、范德华力等各种非共价键相互作用,受到氨基酸序列长度、残基物理化学性质、分子柔韧性[4]及周围的溶剂环境等诸多因素的影响[5]。蛋白质折叠速率 (通常用Kf表示)作为度量蛋白质折叠快慢的一个参数,可用于探究和分析蛋白质的折叠机制。自1998年Plaxo与其伙伴提出接触序 (contact order,CO)方法用于预测Kf以来,到目前为止,已出现了大量的蛋白质折叠速率预测方法。根据对构象熵的不同处理,预测方法大体分成两类:基于蛋白质大小 (如链长度或有效长度)等非特异属性的预测方法,以及基于蛋白质结构中特异相互作用信息的预测方法[6]。前者对于预测序列长度变化较大的蛋白质的折叠速率比较有效,后者则可以改善序列长度变化较小的蛋白质折叠速率的预测效果。据郭等人[7]的统计,截至2006年,已出现了基于结构信息的预测方法,如CO、ΔG(free-energy landscapes)、Leff(effective length)、SSC(secondary structure contact)、ECO(effective contact order)、LRO(long-range order)、TCD(total contact distance)、CTP(chain topology parameter)、Flocal(fraction of local contacts);基于一级序列的预测方法,如HP(helix parameter)、L(chain length)、Pave(average properties of amino acids)、Ω (表示氨基酸属性)等。不难发现,所有这些经验参数中,大多数需要蛋白质的结构信息,而且包含的折叠影响因子单一,加上预测使用的数据量少,使得这些方法并不适用于所有的蛋白质。随后,科学家们渐渐开始将研究对象转向了氨基酸序列与折叠速率的关系,即利用蛋白质一级序列来预测Kf的方法,设计了许多根据序列构成信息及氨基酸性质进行预测的方法。随着蛋白质折叠实验数据的日益增加、结构预测方法的逐步完善[8],以及蛋白质动力学数据库的出现[9~11],用于预测的参数类型逐渐增加,预测方法的精度也得到很大的提升。Harihar等人[12]针对LRO方法,通过更新的“两态”蛋白质数据,优化相关参数,在增大的数据集上得到了比传统LRO方法更好的相关性。近年来,许多包含了多因素变量的预测模型被建立起来,而这些变量之间的相互关系对折叠速率也有着不可忽视的影响。本文从近几年来预测研究的几个主要方向,对新出现的预测方法作了简要综述。
基于链长度的预测方法
一直以来,肽链长度作为一个描述蛋白质大小的非特异性参数,在众多预测方法中都受到重视。首先,Galzitskaya等人[13]认识到链长度在“三态”蛋白质折叠中是主要的速率决定因子。随后,Ivankov等人[14]又提出了基于二级结构预测的“有效长度”指标Leff,表现出与蛋白质折叠速率很好的相关性。再后来,Ivankov等人[15]又用84个蛋白质数据研究了蛋白质形状参数 (旋转半径、折叠与未折叠部分的横截面半径、紧密度参数)与蛋白折叠速率的关系,发现含有链长度信息的那些参数才表现出与蛋白质折叠速率及折叠中间体出现与否的密切关联。
然而,“有效长度”概念的核心在于其反映蛋白质折叠过程信息的有效性,具体定义则可以有不同的形式。最近,Chang等人[16]从最小集的理念出发,通过穷举20种氨基酸类型的所有可能的组合,选出了与蛋白质折叠速率相关性最好的若干个氨基酸类型,用于“有效长度”的定义;该种定义方式不仅与其建立的能量模型吻合,还取得了良好的折叠速率预测能力:在95个蛋白的数据集上,与折叠速率的相关系数达0.84。
氨基酸的构成指标
2006年,Ma等人[17]较早地关注了氨基酸组成与蛋白质折叠速率之间的关系,研究了不同的氨基酸含量及其相对分子量和简并度对折叠的影响,提出了新的预测参数,即组成指标(CI)。CI的定义为:
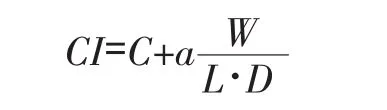
其中,C为某些氨基酸含量之和,W为平均分子量,L为总残基数,D为平均简并度。对37个“两态”和25个“多态”蛋白质进行预测,得到CI与蛋白质折叠速率的皮尔森相关系数 (R)分别为0.73和0.71[17]。通过分析不同折叠动力学类型中各种氨基酸的含量,发现“二态”蛋白和“多态”蛋白在氨基酸组成上存在着差异,且决定折叠速率的氨基酸也不同。在他们接下来的研究中[18],详细列出了不同折叠类型中所含氨基酸的信息:氨基酸F和G多出现于“两态”蛋白质中,而C、H、L和R多出现于“三态”蛋白质中,由此看出,不同类型的氨基酸组成会导致不同的蛋白质折叠动力学类型。将氨基酸构成信息进一步集中在氨基酸的出现频率上,Huang等人[19]在67个蛋白质数据中统计出各序列上不同氨基酸的数目,建立多元回归模型,对蛋白质折叠速率进行预测,“两态”和“多态”蛋白质的相关系数分别为0.78和0.86。通过比较各种氨基酸对应的回归系数来检测折叠反应对残基的敏感性,发现P、N、K、H、R、S、Q、D、G属于折叠促进氨基酸,Y、C、W、L、F、V、I、T、E、A、M则为折叠抑制氨基酸。根据化学基团分类后,氨基基团 (N和Q)、阳离子基团 (K和R)及两性基团 (H)能够促进折叠,而芳香基团 (Y、W、P)和疏水侧链 (L、V、I、A)则会阻碍折叠。考虑到数据集的影响,或许此种分类并不严谨,但在这些结果中,我们已经可以清晰地看到,蛋白质序列上氨基酸的构成信息与蛋白质折叠速率确实存在着很大程度的相关性。
Lin等人[20]综合考虑了序列长度、氨基酸组成、接触序、接触数及二级结构信息,用支持向量机回归模型,对37个“两态”和24个“三态”蛋白进行了预测,得到的相关系数分别为0.81和0.80。在蛋白质数据量相当的情况下 (源于同一篇文献),该方法 (SeqRate)较CI[17]略有提升,由此可以看出,结合多种参数进行预测的方法可能会取得更好的结果,因为考虑的参数越多,就越能覆盖到蛋白质折叠的更多性质,提高相关性。
氨基酸的性质
研究氨基酸的性质有利于对序列上氨基酸比例、氨基酸接触、氨基酸结构偏好及蛋白质折叠顺序等现象进行解释。基于氨基酸性质的折叠速率预测方法在近几年中报道的最多,这表明,分析氨基酸的各种性质对认清蛋白质的折叠有着重要的推动作用。在Gromiha等人[21]的预测方法 (FOLD-RATE)中,对49种氨基酸性质使用多元线性回归,预测77个蛋白质的折叠速率,得到表达式式中,C为常数,a和b为回归系数,P1和P2为氨基酸性质。据称,应用该方法对蛋白质总体进行预测,相关系数为0.96,而如果将蛋白质依据结构分为all-α、all-β和mixed class三类,并对分类后的蛋白质重新进行预测,相关系数分别为0.99、0.97和0.90。其中,mixed class相对较低,这可能是因为该类同时包含了α和β结构,构象更为复杂。在这些氨基酸性质中,结构和热力学特性与all-α蛋白质的折叠速率有很好的相关性;all-β与热力学性质、mixedclass与物理化学性质也都有着较好的相关性。对按结构分类后的蛋白质进行折叠速率预测,结果显示,氨基酸特性与折叠速率有更好的相关性。后来,Huang和Gromiha[22]改进了预测方法,在与FOLD-RATE方法相同的蛋白质和氨基酸性质数据集上,使用了一种新的计算方法——二次响应面模型(quadratic response surface model,QRSM)进行预测,表达式如下:
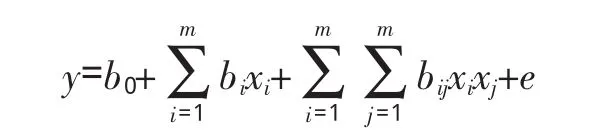
Jiang等人[23]在氨基酸性质的基础上使用混合的序列表示方法,加入氨基酸组成、二级结构特征、序列长度特征等信息,对“两态”、“多态”和“混态” (无法明确分为两态和多态)这三类蛋白质折叠的动力学类型分别建立了线性回归模型 (即prediction of protein folding rates方法,PPFR方法):
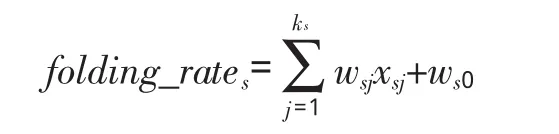
其中,s为集合{“两态”,“多态”,“混态”},xsj为s折叠类型的第j个特征,wsj为s折叠类型第j个特征的回归参数,ws0为拟合常数,而ks表示s折叠类型的特征总数。在62个蛋白中,“两态”和“多态”蛋白折叠速率预测的相关系数均为0.87,“混态”则为0.82。若将此方法 (PPFR)与QRSM用相同的数据作预测比较,QRSM方法得到的相关性要好于PPFR[23],这表明,在本问题中,QRSM二次响应面模型要优于PPFR的线性回归。
对氨基酸性质的更深入理解,建立更好的数学模型,能帮助我们得到更为精确的蛋白质折叠速率预测结果。Gao等人[5]为了分析溶剂可及性、二级结构及残基灵活性对折叠率的影响,同样对三种折叠动力学类型的蛋白质分别建立了线性回归模型 (PFR-AF),对文献[15]中的62个蛋白质进行预测,在“两态”、“多态”和“混态”的蛋白质中分别得到皮尔森相关系数 (R)0.94、0.87和0.84。其中“两态”和“混态”蛋白的相关系数高于用PPFR预测得到的结果,并且,在去除了35%以上相似度的序列冗余后,该方法 (PFR-AF)表现出与折叠速率更好的相关性,相比之下,QRSM和PPFR得到的相关系数则出现明显的下降。从这里可以看出,大多数折叠速率预测方法都存在一定的数据依赖性,不能应对广泛的蛋白质。如果使用序列相似度高的数据,就有可能得到更好的预测值。PFR-AF方法结合了溶剂可及性、残基灵活性及氨基酸组成信息,对一些氨基酸影响折叠的原因作了解释[5],认为:在“两态”蛋白质中,Ala会加速折叠,因其具有较低的构象熵;Ile的增加会延缓两态蛋白的折叠,因为该残基的分支侧链 (branched side chain)会增多潜在的构象;包埋的Pro也会减缓折叠,而暴露的Pro则会加快折叠,因为Pro多存在于蛋白质表面,减少了可能的构象;增加溶剂暴露残基的灵活性会延长折叠时间,主要是由于蛋白质构象的数目增加了。
对于蛋白质折叠过程,氨基酸性质和氨基酸组成共同表现为氨基酸序列的性质。Pred-PFR混合了多种独立的预测项,每一项都基于氨基酸的序列特征,分别对每种特征建立线性回归方程,来预测蛋白质的折叠速率,得R=0.88[24],其回归方程中包含的序列性质有:形成C末端的α-helix的倾向性、形成β-sheet的倾向性、压缩能力、未折叠链的溶剂接触面积、序列长度、有效长度,以及α螺旋、β折叠、coil三种二级结构的比例。Xi等人[25]认为研究序列特征的自相关性有利于认清序列与折叠速率的关系。他们综合考虑了序列的自相关信息、伪氨基酸组成及氨基酸的组成分布等特征,通过遗传算法 (GA),结合多元线性回归 (MLR)和局部懒惰回归 (LLR),对蛋白质折叠速率进行了预测。发现MLR方法下的相关系数为0.93,而LLR方法下得到更高的0.95。将ln Kf分为快、中、慢三个范围,检测20种氨基酸的出现频率,发现赖氨酸 (K)在快速折叠蛋白中的出现频率显著大于慢折叠蛋白 (P<0.005)。若显著性水平P取0.1,则还有N和W会偏向于出现在快速折叠的蛋白质中,而D、V和I则偏向于慢速折叠的蛋白质。
氨基酸的相互作用
2008年,Ouyang和Liang[26]发表了一种基于几何接触和氨基酸序列的蛋白质折叠速率预测方法:几何接触数nα用来表示包裹的非局部接触的数量,定义ln Kf=a+nα×w,其中,a为常数,nα是记录20种残基几何接触数量的20维向量,w是表示相对分子量的20维向量。在80个蛋白的数据集上,预测结果与折叠速率实验值关系显著,相关系数-0.86、-0.86和-0.83分别对应“两态”、“多态”和所有蛋白质。由此可见,无论是对于简单或是复杂的蛋白质,空间包装 (spatial packing)和压缩互作 (zipping interaction)是决定蛋白质折叠速率的重要因素。
网络的概念可以用来描述拓扑和复杂系统的动力学。在Li和Wang[27]的工作中,定义了三种网络:PCNs(蛋白质接触网络)、LINs(长程互作网络)、SINs(短程互作网络)。网络构建以Cα原子作为节点,在每两个节点间建立连接,要求cut-off距离小于0.8 nm。若两节点之间的序列间隔Lcut≥12,则为LINs,否则为SINs。预测结果中,对于“两态”蛋白,PCNs和LINs与ln Kf间只具有很低的相关性 (0.248和-0.118),而SINs却有着较高的正相关系数0.602。可以看出,蛋白质序列上的短程相互作用对影响“两态”蛋白质的折叠速率起着关键作用。
Guo等人[28]为了研究氨基酸残基间的相互作用和氨基酸的序列顺序等信息对折叠速率的影响,采用伪氨基酸组成的方法提取氨基酸序列的位置信息。该方法中,残基间的相关性由残基的疏水值决定,利用蒙特卡洛方法选择最佳预测特征因子,建立线性回归模型进行折叠速率预测。使用91个蛋白质数据,得到的相关系数为0.81[28]。该结果表明,蛋白质序列的疏水氨基酸含量是决定折叠速率的重要因素,且序列顺序信息对蛋白质折叠速率有一定的影响,设计算法时应考虑这种影响,以提高预测精度。最近,Cheng等人[29]进一步利用基于滑动窗口技术的伪氨基酸组成方法,考虑了大量的蛋白质物理化学性质和氨基酸的统计特征,使用非线性支持向量机回归模型对折叠速率进行预测。据称,预测结果与实验数值的相关系数为0.9313。上述研究表明,氨基酸在蛋白质序列中的排列模式对蛋白质的折叠速率有一定的影响。
折叠速率与折叠机制
在蛋白质折叠过程中,二级结构的形成对折叠进程起着决定性的作用。例如。β-sheet在越复杂的蛋白质中折叠得越缓慢[30],helix作为局部结构的代表,能在蛋白质折叠过程中快速形成[2],高helix、高coil含量能促使蛋白质加速折叠[22]。Huang等人[31]假设“两态”和“多态”折叠共享同一模型,则“多态”蛋白质会先缩合为亚稳态的中间体,随后在限速步骤中形成α-helix、turn和β-sheet,该类蛋白质的折叠速率与α-helix和β-sheet结构的长度反向相关;“两态”折叠中,α-helix和turn的较早形成会促进中间体的折叠,因而几乎无法观察到中间体。此模型中,序列长度L表示为L=Lα+Lβ+Lloop,其中Lα、Lβ和Lloop分别表示α、β和loop结构中的残基数。在“两态”折叠中,限速阶段是β-sheet和loop结构的形成,而“多态”中则是α-helix和β-sheet结构的形成。基于该模型,对“两态”和“多态”折叠分别进行预测,公式如下:

对21个“多态”蛋白质进行预测,相关系数为-0.940;对于38个“两态”蛋白质,相关系数为-0.881。为进一步验证二级结构与折叠速率的相关性,作者对单个二级结构和任意两个二级结构的组合与折叠速率的关系进行了比较[31]。对于“多态”折叠,α+β组合的相关系数最高;对于“两态”折叠,β+loop组合的相关系数最高。这在一定程度上证明了该蛋白质折叠模型的可行性。通过此模型发现,在早期中间体中,疏水核心和α-结构的竞争性形成过程会决定折叠的动力学类型,而二级结构的长度则影响着“二态”和“三态”蛋白质的折叠速率,并且,随着相应二级结构长度的增加,蛋白质折叠速率会不同程度地延缓。
序列长度L和基于α-helix的有效序列长度Leff,都曾单独作为特征量被用于折叠速率的预测。Chou和Shen[32]则整合了以上两种特征量及β-sheet性质的影响,建立了线性回归方程,对折叠率进行预测,
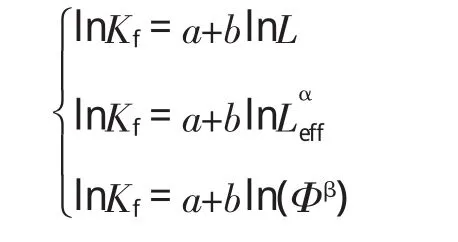
最近,Galzitskaya和Glyakina[34]用一种基于成核机制的理论方法模拟了蛋白质折叠进程,估算了折叠核的大小与自由能障,并在此基础上计算了蛋白质的折叠速率。对84个蛋白质和多肽链,首次得到折叠核大小与实验折叠速率数据的相关系数为-0.57,相关性并不高。但他们同时也发现,估算的自由能障与计算的折叠速率相关性高达0.75,证明了构象熵对折叠速率的重要影响,并且,此成核模型也从物理角度描述了蛋白质的折叠进程。天然蛋白质的形成大致是一个能量递减的过程,“漏斗”状的自由能图景很好地描述了蛋白质折叠到天然状态的过程中熵的整体减少[2,8]。在上述的多种预测方法[5,16,24,26]中,也有从能量观点来解释蛋白质折叠中现象的尝试。虽然目前直接基于能量的折叠速率预测方法并不多,但能量作为分析蛋白质构象的重要参数,对蛋白质折叠速率的预测有着不可忽视的作用。
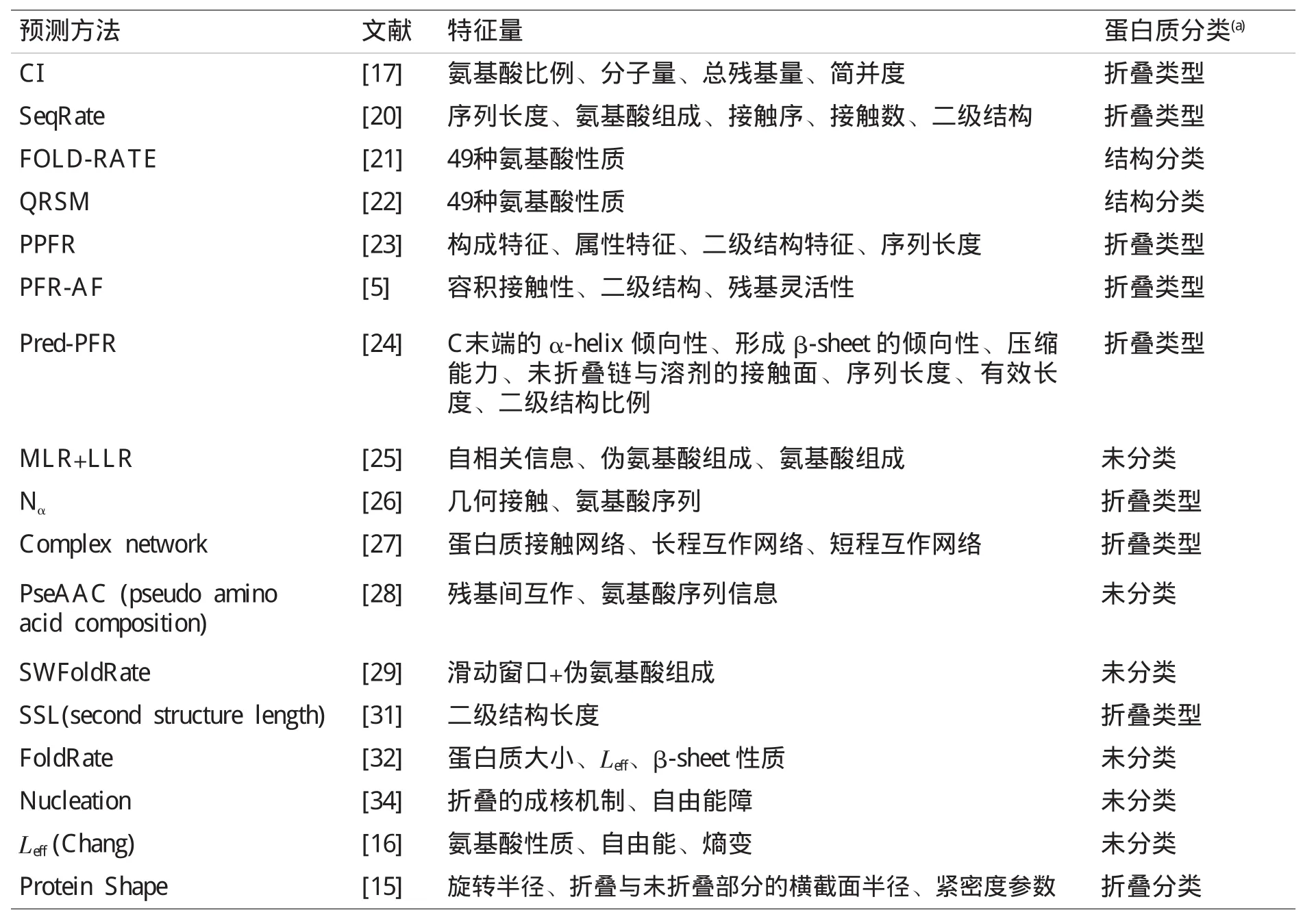
表1 折叠速率预测方法的特征量及蛋白质分类方式Table 1 The features and protein classification schema used in protein folding rate prediction
各种预测方法的归纳与比较
我们对上述主要方法使用的特征量作了整理,结果展现在表1中,接着,用文献[26]中的80个蛋白质数据,对其中有预测网站的6种方法进行了蛋白质折叠速率预测,并计算了这6种方法间的相关性,结果如图1。从图中可以清楚地看出,Pred-PFR、FoldRate和SeqRate两两间的相关性非常高,Pred-PFR和FoldRate的相关性甚至达到了0.99,这可能是因为这三种方法所使用的特征量有很大程度的相似性 (见表1)。近几年来,伴随着预测方法的发展,预测网站逐渐增多,为科学家们进行折叠速率预测和折叠方法研究提供了方便,图2A显示了迄今已有的部分预测网站。在每个预测网站中,都提供了该网站预测方法所使用的蛋白质数据,可供下载使用。自1998年以来,预测方法所使用的蛋白质数据从最初的12个逐渐增加到现在的101个[25],呈增长趋势 (如图2B所示)。蛋白质数据的增加,对检验预测方法的数据依赖性和折叠动力学分类的研究有着重要意义。

蛋白质折叠动力学类型预测
在上述2006年以来的预测方法中,大部分的预测结果都包含了蛋白质的折叠动力学分类 (见表1),且分类后的预测精度都出现了明显的上升。随着蛋白质数据量的增大,将蛋白质进行准确的动力学分类,也成为提升预测结果的重要手段。在“两态”和“多态’两种蛋白质折叠类型中,存在着不同的蛋白质序列组成特征和拓扑复杂性,它们的折叠行为不只依赖于环境条件,更是源于蛋白质的固有性质[18]。基于此,Ma等[18]结合氨基酸含量和序列长度,定义了一种折叠类型预测方法 (Cp):

其中,Length为蛋白序列长度,Csum是显著富含于“多态”折叠类型中的氨基酸的含量之和,a、b和c为三个拟合参数;通过Cp是否大于0,可判定“多态”和“两态”。随后,Huang等人[22]利用10种能正确描述77个蛋白质“两态”和“多态”的氨基酸性质,对蛋白质进行折叠动力学分类,得到了89.6%的准确度。Lin等人[20]发现蛋白质序列长度在预测“多态”折叠速率时是很好的特征量,但却不适用于“两态”;而蛋白质拓扑结构与“两态”蛋白折叠速率的相关性要明显好于“多态”。在一定的数据集上,他们的分类方法得到了80%的准确性。近几年中,虽然出现的预测折叠动力学类型的方法并不多,但是关于折叠动力学类型的讨论却在逐渐增多。对蛋白质进行折叠动力学分类,俨然已经成为认清蛋白质折叠机制的重要步骤。
总结与展望
现阶段出现的蛋白质折叠速率预测方法,或多或少地忽视了蛋白质实际折叠过程中的许多不确定因素,比如二硫键、折叠环境、蛋白质相互作用等。对于每种蛋白质,因为其性质不同,不同方法的预测结果也会不同,且各种性质在折叠中的权重,直接决定着预测方法的好坏。随着蛋白质折叠问题研究的深入,更多的折叠相关参数会被发现,并用于折叠速率的预测。通过预测折叠速率,我们也能反过来解释许多蛋白质折叠中的问题,完善对蛋白质折叠机制的理解,进而推动对蛋白质折叠疾病的认识和治疗药物的研究。
1. 王明,李学周,符兆英.蛋白质错误折叠与蛋白质构象病.延安大学学报 (医学科学版),2009,7(2):12~13.16 Wang M, Li XZ, Fu ZY. Protein misfolding and conformational disease.J Yanan Univ(Med Sci),2009,7(2):12~13.16
2.Lin MM,Zewail AH.Protein folding-simplicity in complexity.Annalen der Physik,2012,524(8):379~391
3.Wang J,Wang W.A computational approach to simplifying the protein folding alphabet.Nat Struct Biol,1999,6(11):1033~1038
4. Huang JT,Tian J.Amino acid sequence predicts folding rate for middle-size two-state proteins.Proteins-Struct Funct Bioinform,2006,63(3):551~554
5.Gao J,Zhang T,Zhang H,Shen S,Ruan J,Kurgan L.Accurate prediction of protein folding rates from sequence and sequence-derived residue flexibility and solvent accessibility.Proteins,2010,78(9):2114~2130
6. Fersht AR.Transition-state structure as a unifying basis in protein-folding mechanisms:Contact order,chain topology,stability,and the extended nucleus mechanism.Proc Natl Acad Sci USA,2000,97(4):1525~1529
7.郭建秀,马彬广,张红雨.蛋白质折叠速率预测研究进展.生物物理学报,2006,22(2):89~95 Guo JX,Ma BG,Zhang HY.Progress in protein folding rate prediction.Acta Biophys Sin,2006,22(2):89~95
8. Dill KA,Maccallum JL.The protein-folding problem,50 years on.Science,2012,338(6110):1042~1046
9.Bogatyreva NS,Osypov AA,Ivankov DN.Kineticdb:A database of protein folding kinetics.Nucleic Acids Res,2009,37(Database issue):D342~346
10.Capriotti E,Casadio R.K-fold:A tool for the prediction of the protein folding kinetic order and rate.Bioinformatics,2007,23(3):385~386
11.Fulton KF,Devlin GL,Jodun RA,Silvestri L,Bottomley SP,Fersht AR, Buckle AM. Pfd:A database for the investigation of protein folding kinetics and stability.Nucleic Acids Res,2005,33:D279~D283
12.Harihar B,Selvaraj S.Refinement of the long-range order parameter in predicting folding rates of two-state proteins.Biopolymers,2009,91(11):928~935
13.Galzitskaya OV,Garbuzynskiy SO,Ivankov DN,Finkelstein AV.Chain length is the main determinant of the folding rate for proteins with three-state folding kinetics.Proteins:Struct Funct Genet,2003,51(2):162~166
14.Ivankov DN,Finkelstein AV.Prediction of protein folding rates from the amino acid sequence-predicted secondary structure. Proc Natl Acad Sci USA, 2004, 101(24):8942~8944
15.Ivankov DN,Bogatyreva NS,Lobanov MY,Galzitskaya OV.Coupling between properties of the protein shape and the rate of protein folding.PloS One,2009,4(8)e6476.DOI:10.1371/journal.pone.0006476
16.Chang L,Wang J,Wang W.Composition-based effective chain length for prediction of protein folding rates.Phys Rev E,2010,82(5 Pt 1):051930)
17.Ma BG,Guo JX,Zhang HY.Direct correlation between proteins'folding rates and their amino acid compositions:An ab initio folding rate prediction.Proteins,2006,65(2):362~372
18.Ma BG,Chen LL,Zhang HY.What determines protein folding type? An investigation of intrinsic structural properties and its implications for understanding folding mechanisms.J Mol Biol,2007,370(3):439~448
19.Huang JT,Xing DJ,Huang W.Relationship between protein folding kinetics and amino acid properties.Amino Acids,2012,43(2):567~572
20.Lin GN,Wang Z,Xu D,Cheng J.Seqrate:Sequencebased protein folding type classification and rates prediction.BMC Bioinform,2010,11 Suppl 3:S1
21.Gromiha MM,Thangakani AM,Selvaraj S.Fold-rate:Prediction of protein folding rates from amino acid sequence.Nucleic Acids Res,2006,34(Web Server issue):W70~74
22.Huang LT,Gromiha MM.Analysis and prediction of protein folding rates using quadratic response surface models.J Comput Chem,2008,29(10):1675~1683
23.Jiang Y,Iglinski P,Kurgan L.Prediction of protein folding rates from primary sequences using hybrid sequence representation.J Comput Chem,2009,30(5):772~783
24.Shen HB,Song JN,Chou KC.Prediction of protein folding rates from primary sequence by fusing multiple sequential features.J Biomed Sci Eng,2009,2:136~143
25.Xi L,Li S,Liu H,Li J,Lei B,Yao X.Global and local prediction of protein folding rates based on sequence autocorrelation information.J Theor Biol,2010,264(4):1159~1168
26.Ouyang Z,Liang J.Predicting protein folding rates from geometric contact and amino acid sequence.Protein Sci,2008,17(7):1256~1263
27.Li HY,Wang JH.Folding rate prediction using complex network analysis for proteins with two-and three-state folding kinetics.J Biomed Sci Eng,2009,2(8):644~650
28.Guo JX,Rao NN,Liu GX,Li J,Wang YH.Predicting protein folding rate from amino acid sequence. Prog Biochem Biophys,2011,37(12):1331~1338
29.Cheng X,Xiao X,Wu ZC,Wang P,Lin WZ.Swfoldrate:Predicting protein folding rates from amino acid sequence with sliding window method.Proteins,2013,81(1):140~148
30.Portman JJ.Cooperativity and protein folding rates.Curr Opin Struct Biol,2010,20(1):11~15
31.Huang JT,Cheng JP,Chen H.Secondary structure length as a determinant of folding rate of proteins with two-and three-state kinetics.Proteins,2007,67(1):12~17
32.Chou KC,Shen HB.Foldrate:A web-server for predicting protein folding rates from primary sequence. Open Bioinformatics J,2009,3:31~50
33.Horwich A.Protein aggregation in disease:A role for folding intermediates forming specific multimeric interactions.J Clin Invest,2002,110(9):1221~1232
34.Galzitskaya OV,Glyakina AV.Nucleation-based prediction of the protein folding rate and its correlation with the folding nucleus size.Proteins,2012,80(12):2711~2727
猜你喜欢
小学生学习指导(低年级)(2020年10期)2020-11-26
数学小灵通(1-2年级)(2020年9期)2020-10-27
新世纪智能(高一语文)(2020年12期)2020-06-01
中国洗涤用品工业(2019年4期)2019-05-11
中成药(2018年1期)2018-02-02
作文大王·低年级(2017年11期)2017-12-05
小学生学习指导(低年级)(2017年12期)2017-11-22
中成药(2017年3期)2017-05-17
科技视界(2016年27期)2017-03-14
动物医学进展(2015年10期)2015-12-07
