聚(3-己基噻吩)作为光谱增感层在有机太阳电池光谱响应增强中的应用
2013-09-17 06:59吴振武韦尚江张东煜明陈立桅马昌期
物理化学学报 2013年8期
吴振武 刘 扬 韦尚江 黄 训 张东煜 周 明陈立桅 马昌期,* 王 华
(1太原理工大学新材料工程技术研究中心,新材料界面科学与工程教育部重点实验室,太原030024;2中国科学院苏州纳米技术与仿生研究所,印刷电子技术研究中心,江苏苏州215123;3中国科学院苏州纳米技术与仿生研究所,纳米生物医学研究部,江苏苏州215123;4中国科学院苏州纳米技术与仿生研究所,国际实验室,江苏苏州215123)
1 引言
有机太阳电池具有成本低、重量轻、可以柔性化、加工方式多样、适于大规模制造等一系列优势,有望作为无机太阳电池的补充,得到了学术界的重视,并且在材料和器件制备工艺上已经有明显进步.1-4目前,基于有机半导体材料的单结光伏器件的光电转换效率已经突破了8%,5而有机多结叠层电池器件的效率则已经突破了10%.4,6作为有机太阳电池的光活性层,有机共轭半导体材料主要承担光吸收、产生激子、激子分离产生自由移动的电荷的作用,其中光吸收是后续系列过程的基础,显得非常重要.目前人们主要通过合成窄带隙的有机共轭分子来提高太阳光的吸收效率.7同无机半导体的宽光谱吸收不同,有机共轭半导体材料的光谱吸收通常为谱带式吸收,因而具有吸收波峰及波谷位置.因此,传统的单一共混异质结结构的太阳电池器件(图1(a))的外量子效率(EQE)会在材料吸收的波谷处较低,不能充分利用这个波段的光子能量.例如,Yu等8报道了(4,8-二烷氧基苯并二噻吩)-(4-氟代噻并[3,4-b]噻吩)共聚合物(PTB4),其薄膜光谱吸收带在600-750 nm,而在400-550 nm处的吸光较弱,利用其与PC61BM制得的光伏器件在400-550 nm处的外量子效率仅为40%左右,在一定程度上限制了器件的整体效率.利用在可见光区有更强吸收能力的C70富勒烯衍生物PC71BM替代吸收较弱的PC61BM可以在一定程度上弥补对光谱响应能力的不足,提高器件整体的光电转换效率.9-10此外,利用光谱具有互补性的两种有机半导体材料来制备叠层电池是解决单结有机太阳电池光谱响应局限的一种常见的手段(图1(b)).11,12例如,Janssen等13利用苯并噻二唑-咔唑交替共聚物(PCPDTBT)与吡咯并吡咯二酮聚合物(PDPP5T)制备了效率达7.5%的双结电池.Yang等14则利用苯并二噻吩-吡咯并吡咯二酮共聚物(PBDTT-DPP)与聚(3-己基噻吩)(P3HT)制备了效率为8.6%的高效叠层电池.相比于单结电池,叠层器件要求前后各结电池的载流子数量和传输过程必须平衡匹配,因此,须对各结电池器件的薄膜厚度等参数进行精确调控,13器件的制备工艺变得更为复杂.

图1 有机太阳电池不同的器件结构示意图Fig.1 Scheme of device structures of various organic solar cells
利用三组份材料共混制备光学活性层是弥补单一材料光谱吸收不足,扩展器件对光谱的响应能力的另外一种重要的方法(图1(c)).15例如You等16利用苯并二噻吩-苯并噻二唑交替共聚物共混实现了7%的光电转换效率,其器件的短路电流(JSC)相对于单节电池提高了40%.Thompson等17利用不同给体材料的组合,可以有效地调节器件的开路光电压及光电转换效率.Brabec18和Chen19等则详细地研究了PCPDTBT及P3HT组合形成三组分共混异质结的器件性能.结果表明,由于PCPDTBT及P3HT在光谱上具有一定的互补性,调节PCPDTBT及P3HT之间的浓度比例,可以有效调节器件对太阳光谱的响应能力,进而在一定程度上提高器件的光电转换效率.相比于叠层电池器件,三元组分共混异质结器件的制备工艺比较简单,但由于不同材料分子能级互相干扰,各组份的浓度需要很精细的调整.16-19
为了克服现有有机太阳电池对太阳光谱吸收能力不足的缺点,本文提出了不同于上述两种器件结构的一种全新器件设计方案.在这一器件结构设计方案中,与光活性层具有光谱互补作用的有机聚合物作为独立的缓冲层置于光活性层与电极之间,利用其光子吸收能力,扩展了器件对太阳光谱的响应能力,起到了光谱增感作用.根据其所起的功能,这一薄膜缓冲层被称为光谱增感层(器件结构如图1(d)所示).本文以P3HT和PBDT-TT-F两种具有光谱互补性能的共轭聚合物为例,详细比较研究了不同厚度的P3HT作为前置光谱增感层对器件的外量子效率及短路光电流的影响,结果证实这一器件结构设计可以实现器件光谱响应能力的增强,是提高有机太阳电池性能的一种全新的设计方案.
2 实验部分
2.1 实验材料
PEDOT:PSS(Clevios PVP Al 4083)购自德国Heraeus公司.PEDOT:PSS旋涂前通过Rephile的RJN1345NH尼龙水相过滤.结构规整P3HT(SMIP3HT,分子量:Mn=5.0×104g·mol-1,多分散指数(PDI)为1.7,规整度(Re)为95%)购自朔纶有机光电科技(北京)有限公司.PBDT-TT-F(Mn=21.9×104g·mol-1,多分散指数PDI=1.31)购买自苏州纳凯科技有限公司.PC61BM(99%)购买自荷兰Solenne B.V.公司.氯苯(色谱纯)购自国药集团.二氯甲烷(分析纯)购自江苏强盛功能化学股份有限公司.
2.2 实验设备
光谱吸收测试是由美国PerkinElmer公司的Lambda 750紫外分光光度计完成的.PBDT-TT-F的循环伏安法测试是由荷兰ECO Chemie公司的Autolab PGSTAT302N电化学工作站完成的.使用的真空镀膜机真空度为5×10-4Pa,氟化锂的蒸镀速率为0.1 nm·s-1,Al的蒸镀速率为0.2-0.3 nm·s-1.器件J-V测试使用美国keithley2400数字源表,模拟太阳光由50 W卤钨灯光源通过HOYA R208449-11060蓝光滤光片和R176892-10047紫外滤光片组合形成,光强经过标准Si探测器及外量子效率EQE校准.EQE测试使用美国Newport的IPCE/QE测试系统.膜厚测试是由Deklak 150台阶仪测试的.
2.3 器件的制备工艺
2.3.1 溶液配制
根据实验需求,共配制如下四份溶液.
A溶液:PBDT-TT-F、PC61BM及添加剂1,8-二碘辛烷(DIO)按m(PBDT-TT-F):m(PC61BM):V(CB):V(DIO)=5.0 mg:7.5 mg:1.0 mL:15 μL配比溶于氯苯(CB)中;
B溶液:P3HT溶于氯苯中,浓度为6 mg·mL-1;
C溶液:PC61BM溶于二氯甲烷中,浓度为6 mg·mL-1;
D溶液:PBDT-TT-F、PC61BM及添加剂1,8-二碘辛烷(DIO)按m(PBDT-TT-F):m(PC61BM):V(DCM):V(DIO)=4 mg:6 mg:1.5 mL:30 μL配比溶于二氯甲烷(DCM)中.
将上述四种溶液置于惰性手套箱内分别密封,在50°C条件下搅拌12 h.
2.3.2 ITO玻璃片清洗
先将刻蚀好的带有图案化的ITO玻璃片用洗洁精清洗,用去离子水洗净擦干后再用丙酮棉擦拭,干燥后依次使用去离子水、丙酮、异丙醇在超声波清洗槽内分别超声清洗.将异丙醇超声清洗后的玻璃片用干燥的氮气枪吹干,将ITO玻璃片转移到真空氧等离子体处理器内,腔内压力为20 Pa,等离子体清洗3 min.
2.3.3 旋涂和退火
等离子处理好的ITO玻璃片放置在旋涂机中,PEDOT:PSS通过45 μm的水相过滤头,滴在ITO玻璃片上,开动旋涂机旋涂速率为3000 r·min-1,旋涂时间为1 min,其厚度大约为35 nm.将旋涂好的PEDOT:PSS ITO玻璃片放在惰性气氛下120°C的热台上烘干15 min,待器件干燥后,让器件自然冷却至室温.
PBDT-TT-F:PC61BM标准器件的制备.将带有PEDOT:PSS的玻璃片旋涂A溶液,旋涂速率为1200 r·min-1,旋涂时间为1 min,薄膜厚度约为85 nm,旋涂完成后器件不需要退火,直接进行下一步的真空镀膜工艺.
P3HT/PC61BM平面异质结器件的制备.将带有PEDOT:PSS的玻璃片旋涂P3HT的B溶液,旋涂速率为600 r·min-1,旋涂时间为1 min,薄膜厚度约为30 nm,然后旋涂PC61BM的C液体,旋涂速率为3000 r·min-1,旋涂时间为1 min,薄膜厚度约为10 nm,旋涂完成后器件不需要退火,直接进行下一步的真空镀膜工艺.
结构为ITO/PEDOT:PSS/P3HT/PBDT-TT-F:PC61BM/LiF/Al的制备.将带有PEDOT:PSS的玻璃片旋涂P3HT的B溶液以不同的旋涂速率旋涂.例如600、1000、1500、3000 r·min-1,旋涂时间为1 min,经台阶仪测试薄膜厚度大约分别为30、25、20、15 nm,旋涂好的器件移至手套箱内惰性气氛下120°C的热台上,退火15 min.将PBDT-TT-F与PC61BM的溶液D旋涂在已经旋涂有P3HT薄膜的器件表面上,旋涂速率为600 r·min-1,旋涂时间为1 min,厚度大约为85 nm,这一步器件不需要热退火.
制备PBDT-TT:PC61BM的参比器件.将带有PEDOT:PSS的玻璃片旋涂D溶液,旋涂速率为600 r·min-1,旋涂时间为1 min,薄膜厚度约为85 nm,旋涂完成后器件不需要退火,直接进行下一步的真空镀膜工艺.
2.3.4 电极蒸镀
将旋涂好的器件转移至真空腔体内,抽真空至1.0×10-4Pa以下,先在器件上镀一层1 nm厚的LiF,然后在器件上镀一层100 nm铝膜作为电极.镀完膜后冷却30 min就可以退掉真空,移出器件,准备测试.
3 结果与讨论
3.1 基于PBDT-TT-F:PC61BM的光伏器件性能
图2为本文所用的聚合物P3HT、PBDT-TT-F和受体材料PC61BM的化学结构.PBDT-TT-F同文献8中报道的PTB4具有相同的化学结构,其主要的光谱吸收位于550-700 nm,对350-550 nm波长的吸收性能较小.与此相对应的,P3HT薄膜的最大吸收位于450-600 nm,与PBDT-TT-F薄膜的光谱吸收形成良好的互补关系(图3(b)).
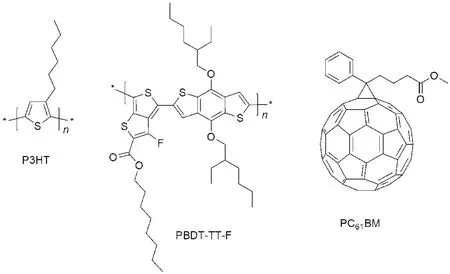
图2 P3HT,PBDT-TT-F和PC61BM的化学结构Fig.2 Chemical structures of P3HT,PBDT-TT-F,and PC61BM
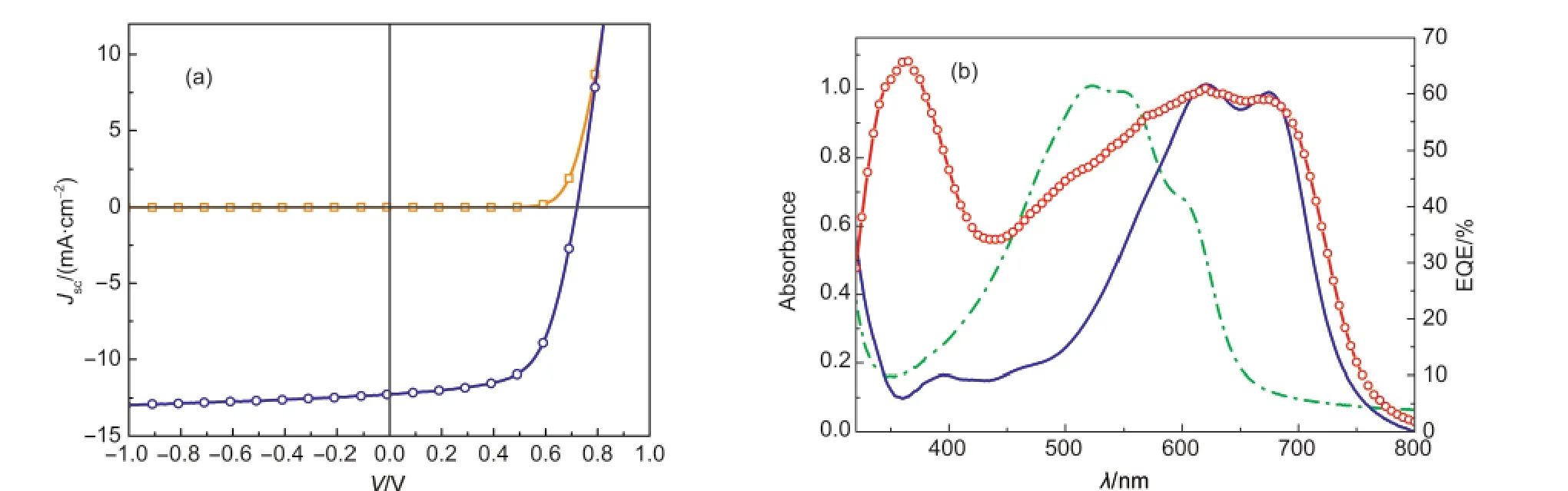
图3 (a)利用PBDT-TT-F作为电子给体材料同PC61BM共混制备的光伏器件的J-V曲线;(b)该器件的外量子效率(EQE)与PDT-TT-F和P3HT固体薄膜的吸收光谱比较Fig.3 (a)J-V curves of a solar cell device with device structure of ITO/PEDOT:PSS/PBDT-TT-F:PC61BM(1:1.5,w/w)/LiF/Al;(b)Comparison of EQE spectrum of the solar cell and the absorption spectra of P3HT and PBDT-TT-F thin films
图3(a)为利用PBDT-TT-F:PC61BM的氯苯溶液(溶液A)制备的结构为ITO/PEDOT:PSS/PBDT-TTF:PC61BM(1:1.5,w/w)/LiF/Al的器件的典型的J-V曲线.通常改变给受体共混的比例也可以明显改变器件的性能,20本例中使用的PBDT-TT-F:PC61BM(1:1.5,w/w)是经过优化后的结果.从图中可以看出,利用PBDT-TT-F制备的单结体相异质结电池的开路光电压(VOC)为0.72 V,短路光电流密度(JSC)为12.26 mA·cm-2,填充因子FF=0.63,器件的光电转换效率(PEC)达到了5.56%,同文献8的研究结果非常接近.从图3(b)器件的EQE中可以看出,器件在350及640 nm处有两个较强的光电转换效率带.其中,350 nm处的光电转换过程主要是源于PC61BM的吸收;而波长在550-700 nm处则主要源于PBDT-TT-F的吸收.从图3(b)的EQE光谱中还可以看出,由于PBDTTT-F在350-550 nm处的吸收较弱,器件在这一波段的量子转换效率较低,在一定程度上限制了器件整体的转换效率.通常在以P3HT:PCBM为活性层的有机太阳电池器件中,热退火处理可以有效改善器件的性能,21然而对于PBDT-TT-F:PCBM为活性层的热退火实验表明,该器件对退火温度比较敏感,在120°C退火1 min,会导致器件性能明显下降.
3.2 利用P3HT作为光谱增感层的器件
3.2.1 二氯甲烷对P3HT薄膜的影响
多层结构有机薄膜光伏器件的全溶液法制备对制备过程中的溶剂选择提出了更高的要求.这是由于上层溶液的溶剂容易发生渗透,破坏下层薄膜的结构.已有文献报道利用溶液法制备P3HT/PC61BM平面异质结的器件,作者声称利用二氯甲烷作为溶剂,不会破坏P3HT层,因而可以制备P3HT/PC61BM平面异质结的器件.22Lee、23Chabinyc24及Dadmun25等通过实验证实,利用二氯甲烷作溶剂制备的初始P3HT/PC61BM平面异质薄膜,经过热退火后可以促进PC61BM向P3HT薄膜层内渗透,从而形成共混异质结界面.
为了明确二氯甲烷是否对P3HT薄膜造成破坏,我们在起始厚度为10 nm左右的P3HT薄膜上滴加二氯甲烷溶剂并以3000 r·min-1旋涂处理两次,然后比较了薄膜处理前后的光谱及表面形貌变化.图4为P3HT薄膜经过二氯甲烷处理前后的光谱吸收变化.从中可以看出,P3HT薄膜的吸收峰形用二氯甲烷处理前后变化不大,说明旋涂二氯甲烷溶剂并不会影响到P3HT薄膜的结晶性能.但薄膜的吸收强度大约降低10%,说明二氯甲烷处理后P3HT薄膜的厚度有大约10%的减少.

图4 利用二氯甲烷处理前后的P3HT薄膜紫外-可见吸收光谱变化Fig.4 UV-Vis absorption spectra of P3HT film before and after DCM treatment
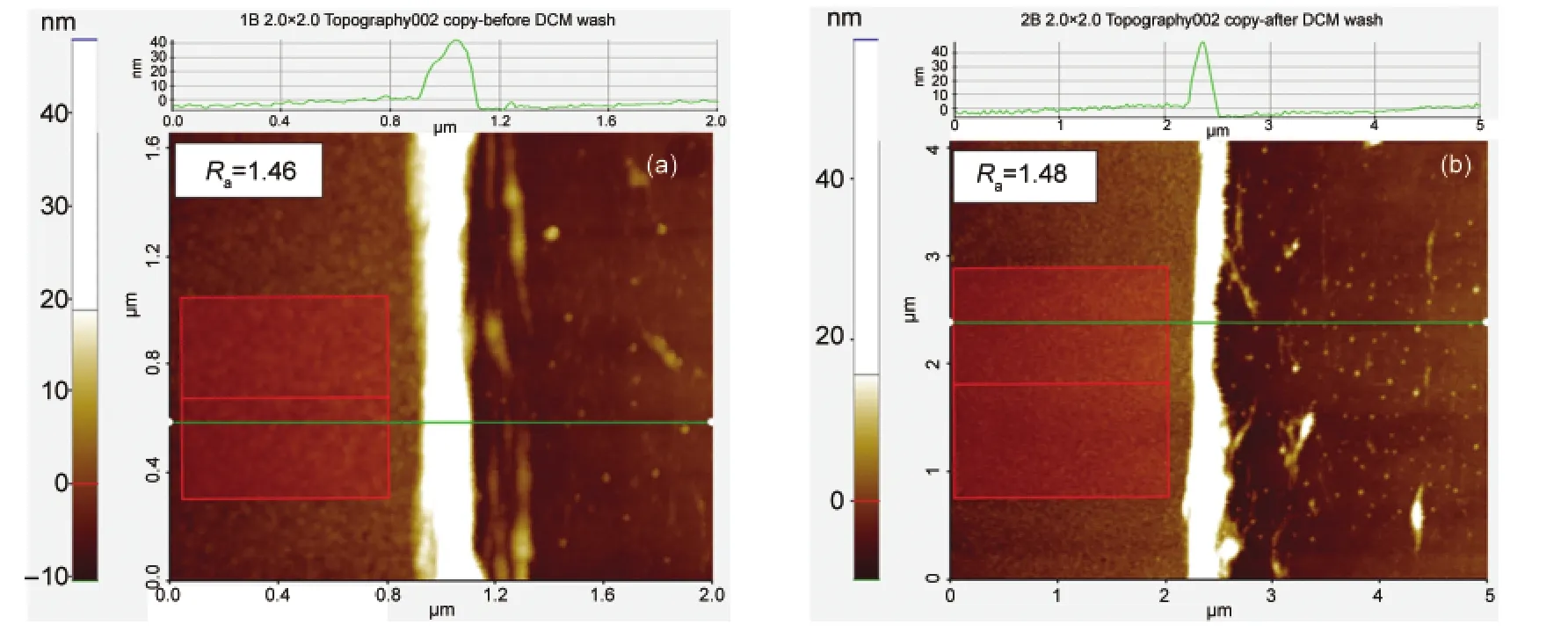
图5 二氯甲烷处理前(a)及处理后(b)的P3HT薄膜划痕处的原子力表面形貌显微图像Fig.5 AFM images of P3HT films at the scratch damaged region before(a)and after(b)DCM treatment
图5为用二氯甲烷处理前后的P3HT薄膜的划痕处的AFM表面形貌图.其中左侧为P3HT薄膜,右侧为划痕区域.从扫描断面估计出P3HT的厚度大约为10 nm左右,同利用台阶仪以及紫外吸收光谱法测得的厚度相近.从图中可以看出,二氯甲烷处理前后P3HT薄膜的表面粗糙度从1.46 nm(图5(a))变为1.48 nm(图5(b)),二者相差很小.这一结果表明,二氯甲烷处理P3HT对其薄膜的厚度及表面形貌影响不大,可以利用DCM作为溶剂实现在P3HT薄膜上沉积光学活性有机层.这为本论文实现在P3HT薄膜上利用溶液法沉积PBDT-TT-F:PC61BM光学活性层提供了一个实验的可行基础.
3.2.2 P3HT层对复合薄膜吸收光谱的影响
在二氯甲烷不破坏P3HT的实验基础上,利用二氯甲烷作为溶剂,制备PBDT-TT-F:PC61BM共混溶液,将其旋涂在厚度不同的P3HT薄膜上,制备了结构为ITO/PEDOT:PSS/P3HT/PBDT-TT-F:PC61BM的系列复合薄膜.图6为具有不同P3HT厚度的P3HT/PBDT-TT-F:PC61B复合薄膜的紫外-可见吸收光谱.作为比较,图中也列出了P3HT/PC61BM及PBDT-TT-F:PC61BM薄膜的吸收光谱.从图中可以看出,PBDTTT-F:PC61BM混合薄膜的主要光谱吸收带在580-720 nm处,其在400-580 nm呈现吸收波谷.而P3HT:PC61BM薄膜的吸收则是在400-600 nm处,与前者形成良好的互补性.P3HT/PBDT-TT-F:PC61BM复合薄膜的吸收可以分为400-600 nm及650-750 nm两个部分.其中400-600 nm处的吸收随着P3HT的厚度的增加而增加,因此可以将这一波段的吸收主要归结于P3HT薄膜的吸收.而600-750 nm处的光谱吸收并不随P3HT的厚度发生明显的改变,因而将这一波段归结于PBDT-TT-F的吸收.从图中可以看出,当引入15-20 nm左右的P3HT时,复合薄膜的吸收呈现出一种理想的平台式宽光谱吸收.
3.2.3 P3HT光谱增感层对器件性能的影响

图6 不同厚度P3HT/PBDT-TT-F:PC61BM混合薄膜的紫外-可见吸收光谱Fig.6 UV-Vis absorption spectra of P3HT/PBDT-TT-F:PC61BM films with different thicknesses
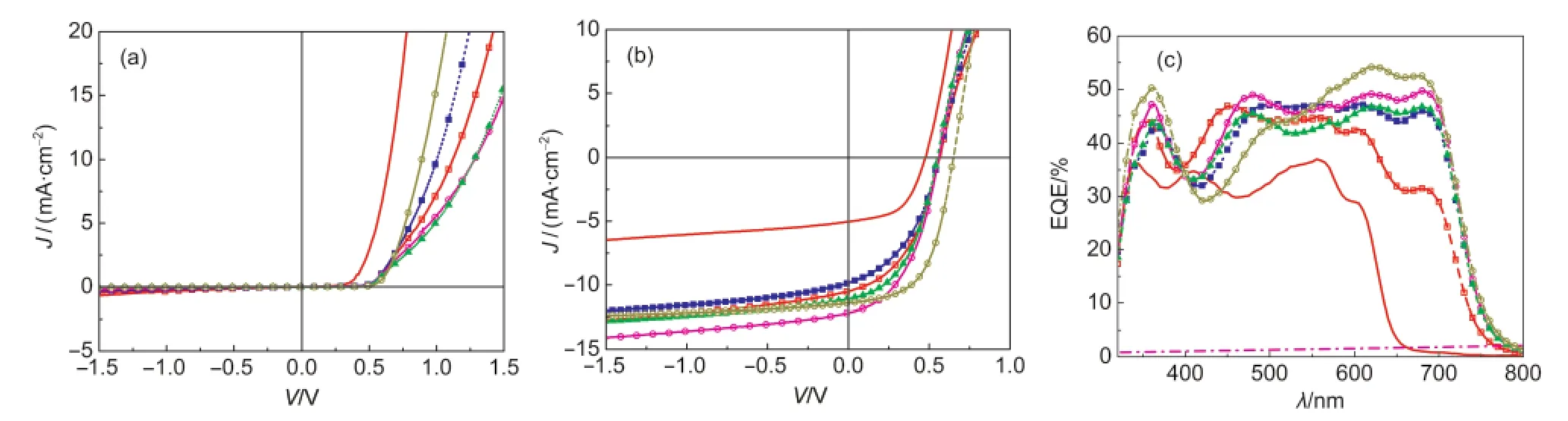
图7 具有不同结构的异质结(BHJ)电池器件在暗态(a)及亮态(b)下的J-V曲线以及器件的EQE谱(c)Fig.7 J-Vcurves(a,indark;b,underillumination)and EQEspectra(c)of variousBHJcelldeviceswith different structures
在上述实验基础上,制备了结构为ITO/PEDOT:PSS/P3HT/PBDT-TT-F:PC61BM/LiF/Al的系列器件.作为比较,也制备了利用二氯甲烷溶液制备的PBDT-TT-F:PC61BM体相异质结电池以及P3HT/PC61BM平面异质结电池.图7为上述系列器件的暗态(a)及亮态(b)曲线,以及器件的外量子效率谱(c).表1列出了测得的不同器件的物理性能参数.从表1中可以看出,P3HT/PC61BM平面异质结电池未经退火时器件的最大光电转换效率为1.26%,接近文献22报道的结果(1.4%).而利用二氯甲烷溶液制备的PBDT-TT-F:PC61BM体相异质结电池器件的开路光电压VOC=0.68 V,短路电流JSC=11.42 mA·cm-2,填充因子FF=0.53,最大光电转换效率为4.12%,略低于利用氯苯溶液制备的器件的性能(参见本文3.1节).这可能是由于二氯甲烷的挥发性较氯苯强,薄膜沉积过程过快而不能形成理想的给受体纳米结构.
从图7(a)中可以看出含有P3HT光谱增感层的器件在暗态下其整流效应较不含P3HT薄膜层的器件差,其开态电流较小.这可能是由于P3HT层的引入,增加了电荷的注入与传输的困难,提高了器件的串联电阻.在光照情况下,含有P3HT薄层的器件的开路光电压(VOC)均为0.60 V左右,比简单的P3HT/PC61BM双层结构器件开路电压(0.51 V)提高了0.09 V,但却比PBDT-TT-F:PC61BM异质结电池的VOC降低了0.08 V.有意思的是,器件的开路光电压同P3HT薄膜的厚度并无明显的依赖关系,这说明在PBDT-TT-F:PC61BM异质结界面产生的空穴很快地都注入到P3HT的HOMO能级,因而器件的开路光电压主要是由P3HT与PC61BM的费米能级所决定.
与开路光电压不同,器件的JSC与P3HT薄膜的厚度有着明显的相关性.当P3HT的厚度为15 nm左右时,器件的JSC为11.08 mA·cm-2,略低于不含P3HT薄膜的器件JSC(11.42 mA·cm-2).而当P3HT的厚度为20 nm左右时,器件的JSC=12.15 mA·cm-2,较不含P3HT层的器件有6%的提高.EQE光谱测试结果表明,在引入P3HT薄层后,器件在400-580 nm处的光谱响应得到了提高.表明P3HT吸收光子形成激子后,激子能够扩散到PC61BM界面并形成有效的电荷分离.P3HT实现光谱增感作用的机制存在以下两种可能:(1)能量转移机制.即P3HT吸收光子形成激子后,通过能量转移将能量传递到PBDTTT-F,之后由PBDT-TT-F与PC61BM之间发生电荷分离;(2)直接电荷分离机制.P3HT薄膜与PBDTTT-F:PC61BM混合薄膜中直接接触的PC61BM形成平面异质结界面,P3HT吸收光子形成的激子在上述界面中形成有效的电荷分离.已有文献26报道P3HT/P3HT:PC61BM复合薄膜中能够形成有效的P3HT/PC61BM平面异质结界面,有利于电荷的分离,支持了上述的直接电荷分离机制.尽管本文的实验尚不能排除上述的能量转移机制,考虑到P3HT薄膜的荧光量子效率低等因素,我们认为,P3HT增感作用主要是通过直接电荷分离机制实现的.有趣的是,尽管文献27报道P3HT激子的扩散长度在8.5 nm左右,本实验结果表明厚度在20 nm左右的P3HT薄膜仍然能够有效地实现激子的分离.这可能是在PBDT-TT-F:PC61BM成膜过程中,还是有少量PC61BM渗透,从而形成P3HT:PC61BM异质结.23从EQE谱图中可以看出,当P3HT的厚度大于20 nm时,器件量子效率整体下降,器件对外输出JSC也从12.15 mA·cm-2下降到10 mA·cm-2左右(图7(c)及表1).器件在红光区域(主要为PBDT-TT-F的吸收)量子效率的降低,可能是由于P3HT薄膜的引入,改变了器件的光程结构,使器件光学活性层偏离了PBDT-TT-F吸收的理想位置;28-30而器件在400-600 nm处(主要为P3HT的吸收)量子效率的下降,则可能是由于P3HT厚度的增加,增加了P3HT激子扩散到PC61BM界面的距离,降低了激子的分离效率.从上述的实验结果中可以看出,厚度小于20 nm的P3HT薄膜可以提高器件对400-600 nm波段光子的响应能力,实现了光谱增感的作用.热退火可以促进PC61BM分子向P3HT薄膜的渗透,有利于P3HT:PC61BM之间形成异质结的增大.然而本实验设计的器件在经过热退火后并不能提升器件性能,这可能是由于PBDT-TT-F:PC61BM异质结对热的敏感所致(参见3.1节).
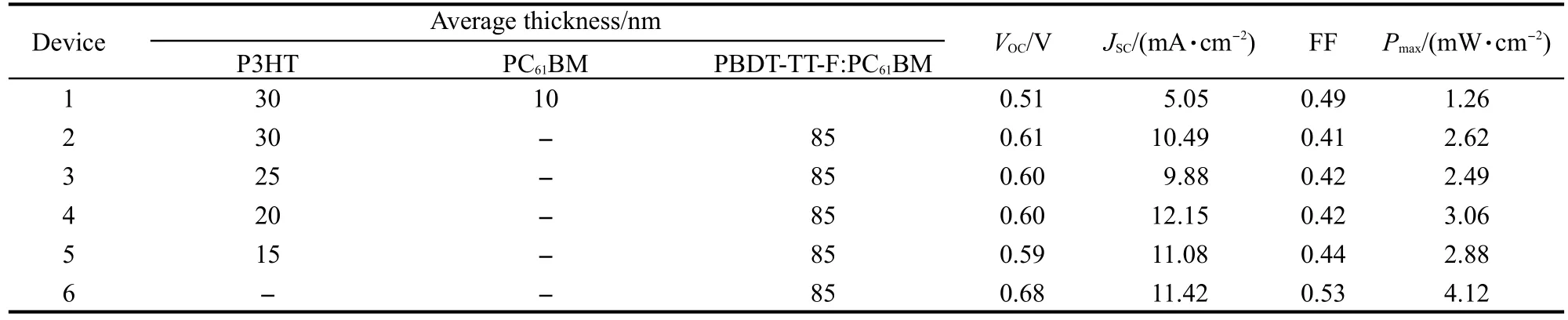
表1 器件的各项参数Table 1 Deviceʹs parameters and performance data
3.2.4 P3HT光谱增感层对器件物理过程的影响
利用循环伏安法测试了PBDT-TT-F及P3HT的氧化还原电位,从中得到PBDT-TT-F及P3HT的最高占据分子轨道(HOMO)/最低未占据分子轨道(LUMO)的能级,其结果与文献8报道结果一致.图8(a)为器件功能薄膜的能级电位示意图.由图可以看出,PBDT-TT-F的光谱带隙为1.8 eV,较P3HT小0.1 eV.PBDT-TT-F的HOMO能级比P3HT低0.1 eV左右,而其LUMO能级比P3HT低0.2 eV.从图8的能级电位中可以看出,P3HT引入器件,P3HT同PBDTTT-F相连,形成一个良好的空穴传输通道,因此,本文所提出的结构设计方案中,P3HT除起到光谱增感的作用外,在器件中还起到了空穴传输层的作用;与此同时,P3HT与光活性共混层中的PC61BM形成异质结界面,这是P3HT在EQE中起到作用的一个基础.
在3.2.3节的讨论中可以看出,含P3HT光谱增感层的器件的开路电压大约为0.60 V,同P3HT:PC61BM共混异质结电池的开路光电压比较一致,而且这一值不随P3HT的厚度变化而变化.从这一结果中可以看出,不论是P3HT/PC61BM异质结界面还是PBDT-TT-F:PC61BM体相异质结界面中受光激发产生的空穴均有效注入到P3HT的HOMO能级,而电子只由PC61BM收集.因此,器件的开路光电压由P3HT和PC61BM的费米能级决定.31-33从图8(a)中可以看出,这个器件实际上是由两个并联的器件组成,其中一个是以P3HT作为空穴传输层的器件,其结构可以理解为ITO/PEDOT:PSS/P3HT(HT)/PBDT-TTF:PC61BM/LiF/Al(HT表示P3HT仅起空穴传输的作用,此处记为D1);而另外一个以P3HT与PBDT-TTF:PC61BM混合层中的PC61BM形成一个平面异质结作为光活性层,其结构可以简化理解为ITO/PEDOT:PSS/P3HT/PC61BM/LiF/Al(记为 D2).由此,器件的等效电路可以表示如图8(b)所示,其中方框内的部分作为子器件D2的等效电路,34其中IL1是PBDT-TT-F:PC61BM有机异质结在光照下产生的光生电流,ID1是PBDT-TT-F:PC61BM异质结自身的漏电流,RS1是PBDT-TT-F:PC61BM异质结自身由于接触和传输产生的阻抗,Ish1是PBDT-TT-F:PC61BM异质结旁路电阻Rsh1产生的漏电.IL2是增感层P3HT/PC61BM有机平面异质结在光照下产生的光生电流,ID2是P3HT/PC61BM有机平面异质结的漏电流,RS2是P3HT/PC61BM有机平面异质结由于接触和传输产生的阻抗,Ish2是P3HT/PC61BM有机平面异质结的旁路电阻Rsh2产生的漏电.从器件能级图(图8(a))和等效电路图(图8(b))中可以看出由于增感层P3HT的引入产生的额外串联电阻RS2也影响到了IL1光电流的输出.
从图8(b)的等效电路图中可以看出,当P3HT的厚度较小的时候(如10 nm左右),由于P3HT薄膜层吸光能力有限,P3HT/PC61BM平面异质结产生的光生电流IL2较小;同时,由于P3HT的引入带来新的串联电阻RS2,因此器件整体的对外输出电流并没有明显的提高.35当P3HT的厚度适中时(本文中20 nm左右),P3HT/PC61BM电流源产生的光生电流增加.尽管RS2也随着厚度的增大而增加,器件最后整体的输出电流有提升.进一步增加P3HT的厚度,一方面提高了RS2,另一方面由于P3HT的光谱吸收,降低了PBDT-TT-F:PC61BM异质结光生电流,因而整体上降低了器件的短路光电流.从上述的分析可以看出,利用有机光谱增感层需后,光谱增感层的厚度直接影响着光生电流IL1与IL2,同时也改变RS2的大小.因此,要实现器件性能整体上的提升,还需要仔细优化P3HT光谱增感层的厚度来平衡IL1、IL2及RS2.
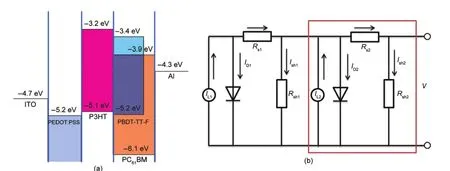
图8 (a)光伏器件的能级结构示意图;(b)光伏器件的等效电路图Fig.8 (a)Energy diagram of the device;(b)equivalent electric circuit of the device
从表1中可以看出,P3HT光谱增感层的引入,除了降低器件的开路光电压外,还降低了器件的填充因子,最终影响到了器件的效率.这是由于P3HT的引入,增加了串联电阻RS2,同时,由于新的异质结界面的引入,增加了电子与空穴的复合几率.我们认为本文中P3HT与PBDT-TT-F:PC61BM薄膜的匹配程度并不是很好,后续的工作可以通过更换材料组合的方式提高器件的效率.因此,根据以上理论分析我们提出,要想发挥光谱增感层的效果,需要一种具有较宽的光学带隙(Eg=2.0 eV)、HOMO能级与PBDT-TT-F相近(EHOMO=-5.0 eV)、具有很高的电荷迁移率及光谱吸收系数的材料,从而能够在较薄光谱增感层(RS2较小)的情况下就产生足够的光吸收补偿(IL2较大),并且保持器件较高的VOC及FF值.
从表1中可以看出,P3HT薄膜的引入可以提高器件的短路光电流.但从上文的分析可以看出,P3HT薄膜作为前置光谱增感层,一方面改变了器件费米能级分布,使得器件的VOC下降;另一方面由于器件串联电阻的增加,导致器件的FF也出现了下降.因此,器件的整体性能并没有得到提升.考虑到P3HT的引入改变了太阳光在器件中的行进路程,影响了PBDT-TT-F的吸收能力(其中一个结果是器件在600-720 nm附近的量子效率降低),因此,我们建议采用具有高空穴迁移率及低费米能级的新型聚合物材料,并将其作为后置光谱增感层将有可能实现器件性能的整体提升.
4 结论
本工作讨论了将P3HT作为光谱增感层,PBDTTT-F:PC61BM作为光活性层的一种新器件结构,通过引入P3HT薄膜作为光谱增感层,弥补了PBDTTT-F:PC61BM作为共混光活性层吸收的不足.由EQE曲线可以看出,P3HT薄膜层的引入,可以提高基于PBDT-TT-F:PC61BM共混异质结电池在400-600 nm处的光谱响应能力.当P3HT厚度为20 nm左右时,器件的短路光电流由11.42 mA·cm-2提高到了12.15 mA·cm-2.通过对器件的等效电路分析,我们提出,理想的光谱增感薄膜需要有与光活性层的主体吸光材料具有良好的光谱互补性,同时其HOMO能级与光学活性层主体吸光材料相近,具有很高的电荷迁移率及光谱吸收系数的材料.
致谢: 本文作者衷心感谢中国科学院化学研究所有机固体实验室王吉政研究员的讨论与指导;感谢苏州纳米技术与仿生研究所印刷电子学部骆群博士在台阶仪测量膜厚方面的帮助;感谢苏州大学材料与化学化工学部的李耀文老师和李超、张盼同学在循环伏安法测试过程中的帮助.
(1)Mayukh,M.;Jung,I.H.;He,F.;Yu,L.J.Polym.Sci.B:Polym.Phys.2012,50(15),1057.doi:10.1002/polb.23102
(2) Kumar,P.;Chand,S.Prog.Photovoltaics Res.Appl.2012,20(4),377.doi:10.1002/pip.v20.4
(3) Brabec,C.J.;Gowrisanker,S.;Halls,J.J.M.;Laird,D.;Jia,S.;Williams,S.P.Adv.Mater.2010,22(34),3839.doi:10.1002/adma.200903697
(4) Li,G.;Zhu,R.;Yang,Y.Nat.Photonics 2012,6(3),153.doi:10.1038/nphoton.2012.11
(5)He,Z.;Zhong,C.;Su,S.;Xu,M.;Wu,H.;Cao,Y.Nat.Photonics 2012,6(9),591.
(6)Green,M.A.;Emery,K.;Hishikawa,Y.;Warta,W.;Dunlop,E.D.Prog.Photovoltaics Res.Appl.2013,21(1),1.
(7)Ye,H.Y.;Li,W.;Li,W.S.Chin.J.Org.Chem.2012,32(2),266.[叶怀英,李 文,李维实.有机化学,2012,32(2),266.]doi:10.6023/cjoc1104062
(8) Liang,Y.;Feng,D.;Wu,Y.;Tsai,S.T.;Li,G.;Ray,C.;Yu,L.J.Am.Chem.Soc.2009,131(22),7792.doi:10.1021/ja901545q
(9) Chen,H.Y.;Hou,J.;Zhang,S.;Liang,Y.;Yang,G.;Yang,Y.;Yu,L.;Wu,Y.;Li,G.Nat.Photonics 2009,3(11),649.doi:10.1038/nphoton.2009.192
(10) Liang,Y.;Xu,Z.;Xia,J.;Tsai,S.T.;Wu,Y.;Li,G.;Ray,C.;Yu,L.Adv.Mater.2010,22(20),E135.
(11)Ameri,T.;Dennler,G.;Lungenschmied,C.;Brabec,C.J.Energy Environ.Sci.2009,2(4),347.doi:10.1039/b817952b
(12) Sista,S.;Hong,Z.;Chen,L.M.;Yang,Y.Energy Environ.Sci.2011,4(5),1606.doi:10.1039/c0ee00754d
(13) Gevaerts,V.S.;Furlan,A.;Wienk,M.M.;Turbiez,M.;Janssen,R.A.J.Adv.Mater.2012,24(16),2130.doi:10.1002/adma.201104939
(14) Dou,L.;You,J.;Yang,J.;Chen,C.C.;He,Y.;Murase,S.;Moriarty,T.;Emery,K.;Li,G.;Yang,Y.Nat.Photonics 2012,6(3),180.doi:10.1038/nphoton.2011.356
(15) Chen,Y.C.;Hsu,C.Y.;Lin,R.Y.Y.;Ho,K.C.;Lin,J.T.ChemSusChem 2013,6(1),20.doi:10.1002/cssc.201200609
(16)Yang,L.;Zhou,H.;Price,S.C.;You,W.J.Am.Chem.Soc.2012,134(12),5432.doi:10.1021/ja211597w
(17)Khlyabich,P.P.;Burkhart,B.;Thompson,B.C.J.Am.Chem.Soc.2012,134(22),9074.doi:10.1021/ja302935n
(18)Ameri,T.;Min,J.;Li,N.;Machui,F.;Baran,D.;Forster,M.;Schottler,K.J.;Dolfen,D.;Scherf,U.;Brabec,C.J.Adv.Eng.Mater.2012,2(10),1198.doi:10.1002/aenm.v2.10
(19) Chang,S.Y.;Liao,H.C.;Shao,Y.T.;Sung,Y.M.;Hsu,S.H.;Ho,C.C.;Su,W.F.;Chen,Y.F.J.Mater.Chem.A 2013,1(7),2447.doi:10.1039/c2ta00990k
(20) Li,D.;Liang,R.;Yue,H.;Wang,P.;Fu,L.M.;Zhang,J.P.;Ai,X.C.Acta Phys.-Chim.Sin.2012,28(6),1373.[李 丹,梁 然,岳 鹤,王 鹏,付立民,张建平,艾希成.物理化学学报,2012,28(6),1373.]doi:10.3866/PKU.WHXB201204061
(21)Zhuo,Z.L.;Zhang,F.J.;Xu,X.W.;Wang,J.;Lu,L.F.;Xu,Z.Acta Phys.-Chim.Sin.2011,27(4),875.[卓祖亮,张福俊,许晓伟,王 健,卢丽芳,徐 征.物理化学学报,2011,27(4),875.]doi:10.3866/PKU.WHXB20110414
(22)Ayzner,A.L.;Tassone,C.J.;Tolbert,S.H.;Schwartz,B.J.J.Phys.Chem.C 2009,113(46),20050.doi:10.1021/jp9050897
(23)Lee,K.H.;Schwenn,P.E.;Smith,A.R.;Cavaye,H.;Shaw,P.E.;James,M.;Krueger,K.B.;Gentle,I.R.;Meredith,P.;Burn,P.L.Adv.Mater.2011,23(6),766.doi:10.1002/adma.201003545
(24)Treat,N.D.;Brady,M.A.;Smith,G.;Toney,M.F.;Kramer,E.J.;Hawker,C.J.;Chabinyc,M.L.Adv.Eng.Mater.2011,1(1),82.doi:10.1002/aenm.201000023
(25)Chen,H.;Hegde,R.;Browning,J.;Dadmun,M.Phys.Chem.Chem.Phys.2012,14(16),5635.doi:10.1039/c2cp40466d
(26)Liang,C.W.;Su,W.F.;Wang,L.Appl.Phys.Lett.2009,95(13),133303.doi:10.1063/1.3242006
(27)Shaw,P.E.;Ruseckas,A.;Samuel,I.D.W.Adv.Mater.2008,20(18),3516.doi:10.1002/adma.200800982
(28) Dennler,G.;Scharber,M.C.;Brabec,C.J.Adv.Mater.2009,21(13),1323.doi:10.1002/adma.v21:13
(29)Kim,J.Y.;Kim,S.H.;Lee,H.H.;Lee,K.;Ma,W.;Gong,X.;Heeger,A.J.Adv.Mater.2006,18(5),572.
(30) Gilot,J.;Barbu,I.;Wienk,M.M.;Janssen,R.A.J.Appl.Phys.Lett.2007,91(11),113520.doi:10.1063/1.2784961
(31)Yamamoto,S.;Orimo,A.;Ohkita,H.;Benten,H.;Ito,S.Adv.Eng.Mater.2012,2(2),229.doi:10.1002/aenm.v2.2
(32)Kröger,M.;Hamwi,S.;Meyer,J.;Riedl,T.;Kowalsky,W.;Kahn,A.Org.Electron.2009,10(5),932.doi:10.1016/j.orgel.2009.05.007
(33) Zhang,M.;Wang,H.;Tang,C.Appl.Phys.Lett.2011,99(21),213506.doi:10.1063/1.3664406
(34) Li,H.;Zhang,Z.G.;Li,Y.;Wang,J.Appl.Phys.Lett.2012,101(16),163302.doi:10.1063/1.4761246
(35) Gu,J.H.;Zhong,Z.Y.;He,X.;Sun,F.L.;Chen,S.B.J.South-Central Univ.Nationalities(Nat.Sci.Edition)2009,28(1),57.[顾锦华,钟志有,何 翔,孙奉娄,陈首部.中南民族大学学报(自然科学版),2009,28(1),57.]
猜你喜欢
化学工程师(2022年3期)2022-04-19
北方论丛(2021年2期)2021-05-22
上海化工(2021年2期)2021-04-23
焊接(2016年7期)2016-02-27
北京信息科技大学学报(自然科学版)(2016年6期)2016-02-27
云南师范大学学报(自然科学版)(2015年5期)2015-12-26
科技与企业(2015年20期)2015-10-21
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
中国舰船研究(2015年2期)2015-02-10
航天返回与遥感(2014年4期)2014-07-31