
p-π共轭分子的氨基面外弯曲振动模的拉曼光谱
2013-09-17 06:58:46于利娟吴德印田中群
物理化学学报 2013年8期
陶 莎 于利娟 吴德印 田中群
(厦门大学化学化工学院化学系,固体表面物理化学国家重点实验室,福建厦门361005)
1 引言
弱相互作用是一类普遍存在的作用,其中氢键作用占有重要的地位.在生物分子体系中,含有芳香氨基基团的许多分子与其环境形成氢键作用,但这类研究体系往往较为复杂,难于进行系统研究.近些年来,苯胺分子已经成为一个很好的模型分子.1-5前人已对苯胺分子与其它小分子的红外光谱进行测量,通过分析氨基伸缩振动谱峰的变化探讨分子间氢键作用.如Nakanaga等6-9用红外光谱研究了苯胺与一系列小分子(如H2O、NH3、N2和CO等)之间的氢键作用,发现苯胺分子的氨基与小分子的氮或氧原子形成氢键,其导致氨基的N―H伸缩振动发生几十个波数的红移.然而,人们目前普遍关注的是这种弱相互作用对高频区谱峰的影响,很少有人关注氢键作用对氨基的面外弯曲振动模谱峰(ωNH2)的影响.实际上氨基上氮与氢给体形成的氢键作用更强,10,11其引起频率的位移更大,谱峰强度变化也更显著.
拉曼光谱是一种非弹性散射光谱,它以单色性很好的激光作为光源,能够提供化学和生物分子结构的指纹信息,广泛用于无损检测和分子识别技术.在采用拉曼光谱研究苯胺及苯胺类衍生物分子与银表面作用的实验中,氨基面外弯曲振动模的信号相对于分子发生显著的变化.12-14当苯胺分子吸附在银表面上,人们常在900-1000 cm-1区间观测一个很强且很宽的拉曼谱峰,该峰被认为来自于苯环的呼吸振动或氨基的摇摆振动.15-17最近,Zhao等18用密度泛函理论计算了苯胺分子吸附在币族金属表面的拉曼谱峰,观察到了ωNH2振动模的频率明显蓝移,同时其拉曼信号显著增强.他们认为ωNH2振动频率的蓝移是由于沿着该振动模的势能曲线由双势阱转化单势阱,同时势能曲线的曲率增大.该振动模的拉曼强度显著增强可以解释为伴随着ωNH2振动氮原子的杂化在sp2和sp3之间变化,导致电子密度的重新分布,从而极化率导数显著增大.
在本文中,我们选择了乙烯胺和苯胺分子作为研究对象,用密度泛函理论分析这两个分子与水以及与金属表面作用对分子内p-π共轭效应及拉曼光谱的影响,研究分子间的弱相互作用与氨基面外弯曲振动的关系,进一步解释氨基的面外弯曲振动拉曼信号异常产生的原因.
2 理论计算方法和模型
为了研究弱相互作用对氨基的面外弯曲振动模的影响,我们构造了分子与水簇作用以及分子与银簇作用的两种理论模型.选择乙烯胺分子作为探针分子,主要是该分子涉及分子内p-π作用,且前人对其分子本身的振动光谱有详细研究.19-21首先,对于乙烯胺和苯胺分子与水作用,如图1所示,我们主要考虑水以O―H键与氨基氮原子形成氢键.随水分子数增加,氨基的N―H键也与水分子的氧成氢键作用.因氢键作用很依赖于介质环境,我们用溶质密度(SMD)的溶剂化模型,22其考虑氢原子的影响和非静电作用项,能很好地预测溶剂化的吉布斯自由能.我们选择水作为溶剂,取水体相的相对介电常数值为78.3.
其次,为了研究分子在银纳米结构表面的吸附,选取银簇模拟金属表面的吸附活性位,研究了两个分子与银簇的作用对氨基的面外弯曲振动模的影响.在分子-银簇模型中,分子与银簇的作用主要是通过氨基孤对轨道形成配位作用.因为分子-银簇的相互作用依赖于吸附位和金属簇的大小,因此我们选择了不同大小的银簇Agn(n=1,2,3,4)模拟银表面可能的活性位.依据成键规则,如能量相近、轨道对称性的匹配和最大重叠,选择的银簇不仅可以反映表面活性位的电子结构特性,而且也能较好地描述乙烯胺和苯胺与银簇作用的成键作用.在以前研究中,该模型合理地模拟了吡啶分子吸附在粗糙金属表面的拉曼光谱.23
为了考查不同密度泛函对计算结果的影响,采用不同的杂化密度泛函B3LYP、X3LYP和M06进行了理论计算.本文所有密度泛函理论计算由Gaussian 09程序24得到.在计算中对分子中的原子采用全电子基组6-311+G(d,p),其为C、N、O和H原子提供了极化函数,为C、N和O原子提供了弥散函数.25,26对银原子采用相对论赝势和相应基组LANL2DZ描述金属内壳层的电子和价层电子.27,28基于上述方法和基组,我们计算了苯胺和乙烯胺分子本身的面外弯曲振动的频率和拉曼散射因子.同时,我们也用B3LYP方法计算分子-水簇以及分子-银簇的成键能和拉曼光谱.计算分子与金属之间的成键能定义为,其中n代表银簇中银原子的个数,EMOL-Mn代表复合物的能量,EMOL和EMn分别代表分子和银簇的能量.在成键能计算中,也用基组重叠误差(BSSE)校正方法29,30来校正成键能.还利用自然成键轨道理论(NBO)31,32分析分子间的轨道作用能.

图1 在B3LYP/6-311+G(d,p)水平下计算的气相中苯胺(a)和乙烯胺(b)分子分别与一个、三个和五个水分子的水簇作用的优化结构Fig.1 Optimized structures of aniline(a)and vinylamine(b)interacting with one,three,and five water molecules in the gas phase calculated at the B3LYP/6-311+G(d,p)level
采用公式(1)计算了不同振动模的微分拉曼散射截面,其对应于单个分子第i个振动模的拉曼强度,33
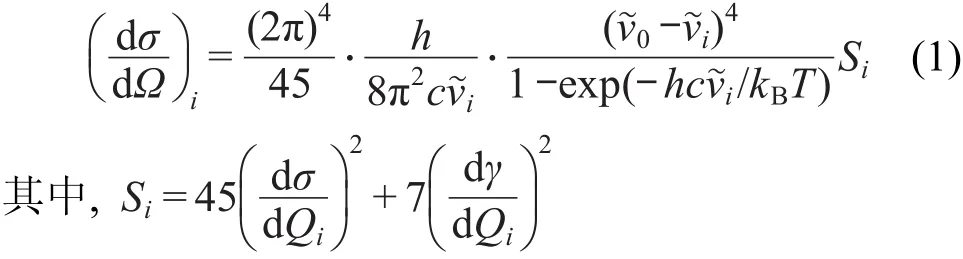
式中,h、c、kB和 T分别为 Planck常数、光速、Boltzmann 常数和Kelvin温度. ν͂0和 ν͂i分别为激发光频率(cm-1)和第i个振动模的振动频率(cm-1).为了与实验拉曼谱进行比较,我们也对200-1800 cm-1波数的密度泛函计算谐频率采用0.981进行校正.Si为拉曼散射因子.dα/dQi和dγ/dQi分别为极化率的各向同性部分和各向异性部分对第i个简正坐标的导数.在理论计算拉曼光谱中,对计算微分拉曼散射截面进行Lorentzian展开,采用最大高度半高宽为10 cm-1,激发光波长为514.5 nm.
3 计算结果与讨论
3.1 苯胺和乙烯胺分子的拉曼光谱
我们利用不同的杂化密度泛函B3LYP、X3LYP和M06的理论计算方法和6-311+G(d,p)基组计算了甲胺、乙烯胺和苯胺分子的面外弯曲振动的频率和强度,比较不同烃基与氨基相连时面外弯曲振动谱性质的变化.表1给出三种不同理论方法计算的面外弯曲振动模的频率和拉曼散射因子.结果表明计算方法对结果影响不大,与甲胺的ωNH2频率相比,乙烯胺和苯胺的频率明显减小,并且这两者较接近.这主要是由于在甲胺分子中氨基是sp3杂化,而在乙烯胺和苯胺分子内存在p-π共轭效应,氨基上的孤对与乙烯基或苯环共轭导致其倾向于sp2杂化性质.由此可见氨基面外弯曲振动模的频率与氨基的杂化性质紧密相关.在氨分子中,其伞式振动频率是958 cm-1.34甲基取代氨的一个氢后,三种泛函预测甲胺的ωNH2频率为808和801 cm-1,其略大于实验观测值(780 cm-1).35在乙烯胺和苯胺中,由于p-π共轭作用,理论计算谐振动频率小于甲胺中相应的频率,而实验观测的频率分别为570和660 cm-1,19,36其大于理论计算谐振动频率.这主要是观测频率不是振动基频,而是由于该振动模均具有显著的非谐性,在自由分子中具有双极小势阱,因此观测频率为泛频.37Rauhut和Pulay38建议其谐振动频率为541 cm-1,与表1计算频率接近.
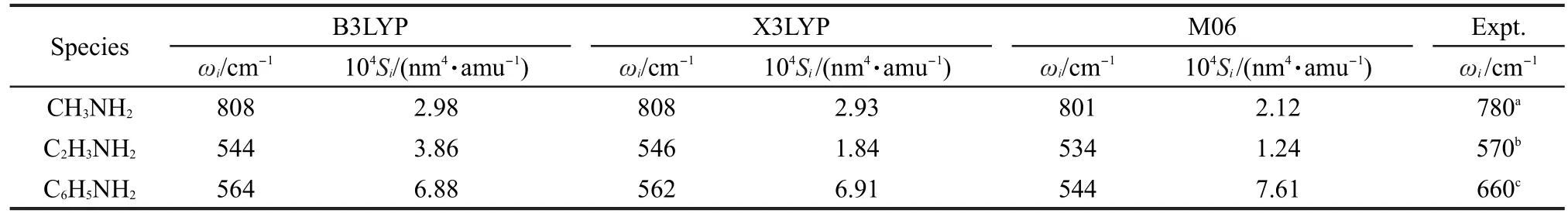
表1 用不同密度泛函理论方法计算的甲胺、乙烯胺和苯胺的氨基面外弯曲振动的拉曼频率(ωi)和拉曼强度(Si)Table 1 Vibrational Raman frequencies(ωi)and intensities(Si)of ωNH2calculated by different density functional theory methods for methylamine,vinylamine,and aniline
图2是采用密度泛函理论计算乙烯胺和苯胺分子内涉及p-π共轭作用的轨道图.在氨基与乙烯基和苯基形成C―N σ键的同时,氨基的孤对轨道也与π轨道发生作用.在乙烯胺分子中,我们发现氨基的孤对轨道主要与乙烯基的π成键轨道作用,形成乙烯胺分子中的一个成键轨道和一个反键轨道.在苯胺分子中,氨基的孤对与苯基的两个π成键轨道相互作用,形成苯胺分子中三个与p-π共轭作用相关的分子轨道.在这两个分子中,上面的分析表明轨道作用主要发生在占据轨道之间,未占据的π反键轨道不产生明显影响.

图2 氨基的孤对轨道分别与乙烯基和苯基的π轨道作用形成乙烯胺和苯胺的p-π共轭轨道的轨道能级图Fig.2 Plots of p-π conjugated orbitals in vinylamine and aniline formed from the lone pair orbital in the amino group interacting with the π orbitals in vinyl and phenyl groups
3.2 氢键作用对氨基面外弯曲振动模的影响
图1 中也给出苯胺和乙烯胺分子以氨基与不同大小的水簇作用的优化结构,它们将用于研究氢键作用对分子中氨基面外弯曲振动模的拉曼信号的影响.图中标出了与分子间氢键相关的N…H―O和O…H―N的氢键键长,括号中为采用溶剂化SMD模型计算的结构参数.比较N…H―O氢键结构的变化可以看出,在苯胺和乙烯胺中随着水分子数目的增加,N…H的距离均逐渐变短.用NBO分析计算氨基氮孤对与O―H键反键轨道作用能,得到在苯胺复合物中其分别为18.07、32.72和64.02 kJ·mol-1,而在乙烯胺复合物中其依次为17.24、31.46和60.96 kJ·mol-1.表明N…H―O氢键随水分子数目增大该氢键作用增强.这也与表2计算的分子之间的氢键的键能越来越大相一致.含有三个水分子和五个水分子的氢键复合物,其N―H…O氢键的H…O原子距离随水分子数增加而增长.这说明氨基的氮原子与水分子的氢原子之间的氢键作用随水分子数增加会变得更加稳定.
图3是在B3LYP/6-311+G(d,p)水平上计算分子-水簇的拉曼光谱.图中标出了含有氨基面外弯曲振动贡献的谱峰,其中用*标出的为亚甲基面外弯曲振动的谱峰.图3(A,C)是在气相中计算的拉曼光谱.从图上可以看到,不论是苯胺还是乙烯胺,其氨基的面外弯曲振动模均依赖于氨基周围水分子的数目,与其分子本身的拉曼光谱(表1)相比,频率有明显的蓝移,强度有明显的增大.如图3(A)所示,当苯胺与不同水簇作用时,在700-1000 cm-1的波数范围内均出现三个包含ωNH2振动坐标的谱峰.通过对振动谱峰的分析,表2结果表明它们来自于面外弯曲坐标与环呼吸振动坐标或C―H面外弯曲坐标的耦合.随着水簇的增大,三个谱峰的频率均有蓝移的趋势.对于乙烯胺与水分子作用,如图3(C)所示,ωNH2的频率也随着水簇的增大发生显著蓝移.在这种情况下,对应于ωCH2的拉曼谱峰位于838 cm-1,其信号在频率和强度上几乎保持不变,可见氨基与水簇的作用对它的影响较小.
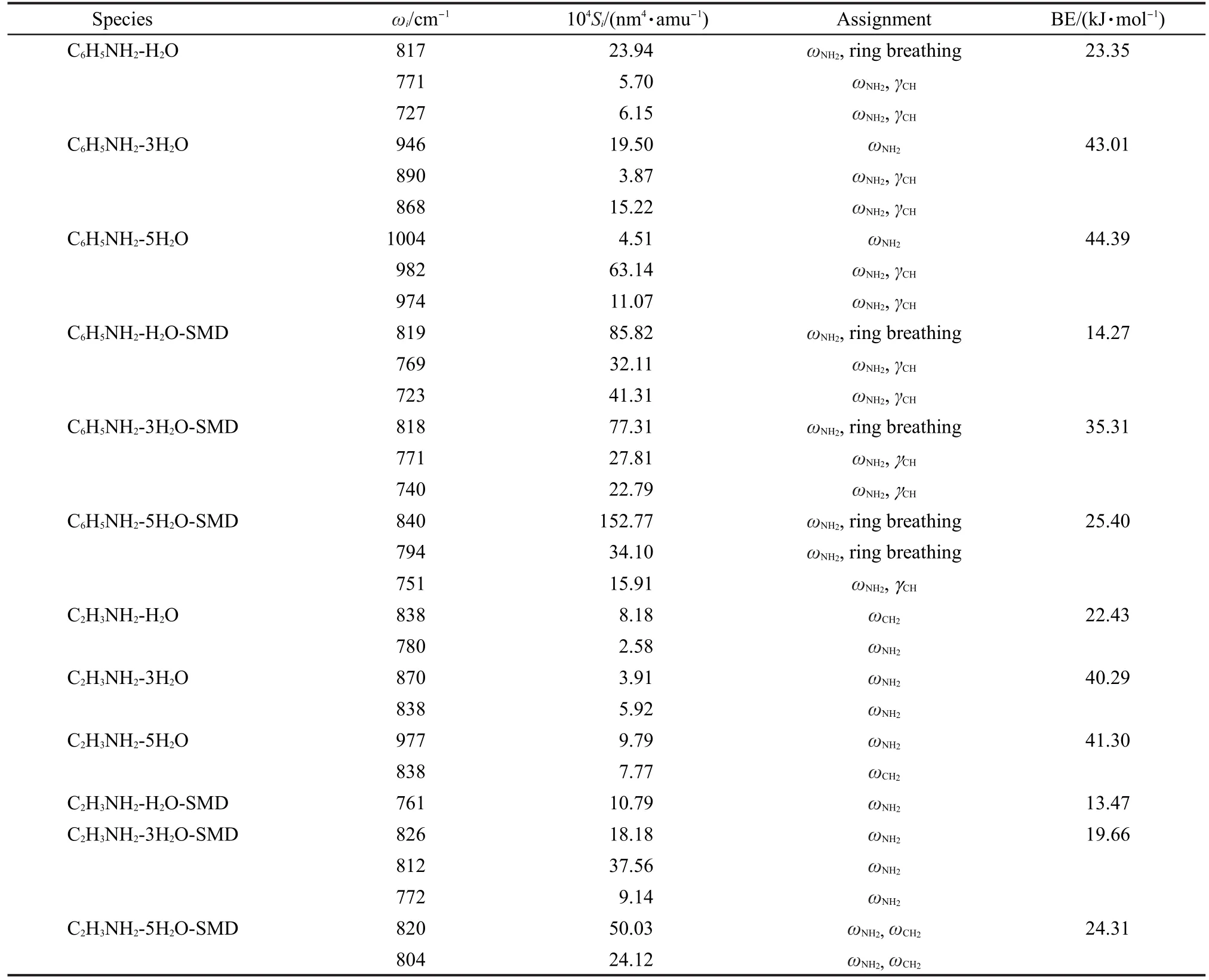
表2 在气相和溶剂化下理论计算苯胺、乙烯胺与水簇复合物的包含面外弯曲振动模的频率(ωi)、拉曼强度(Si)、氢键键能(BE)以及振动模的归属Table 2 Calculated frequencies(ωi),Raman activity(Si),hydrogen bonding energy(BE),and vibrational assignment of ωNH2for C6H5NH2-nH2O and C2H3NH2-nH2O complexes(n=1,3,5)in the gas phase as well as that with SMD model
为了模拟水体相中分子拉曼光谱的变化,采用SMD模型考虑溶剂化效应在氢键作用上的影响.由图1给出氢键结构参数可见,考虑溶剂化后,分子与H2O作用的N…H―O氢键的距离仍随水分子数目增加保持缩短趋势.10,11相对于气相情况,在两个分子与一个和三个水分子形成的簇中,溶剂化效应导致N…H―O氢键的距离均缩短,而当两个分子与五个水分子形成的簇中,N…H―O氢键的距离略有增长.在考虑溶剂化条件下,采用NBO分析计算了N…H―O氢键的轨道作用能,得到在苯胺复合物中其分别为28.20、34.85和41.84 kJ·mol-1,而在乙烯胺复合物中其依次为27.24、31.88和38.79 kJ·mol-1.与气相NBO计算的轨道作用能相比,前两者是增强的,后者是减小的.这与我们计算的氢键距离的变化相吻合,表明虽然溶剂化作用导致贡献到氢键作用的静电作用能减小,但轨道作用相对增强,并且在三个复合物中趋向互相接近.
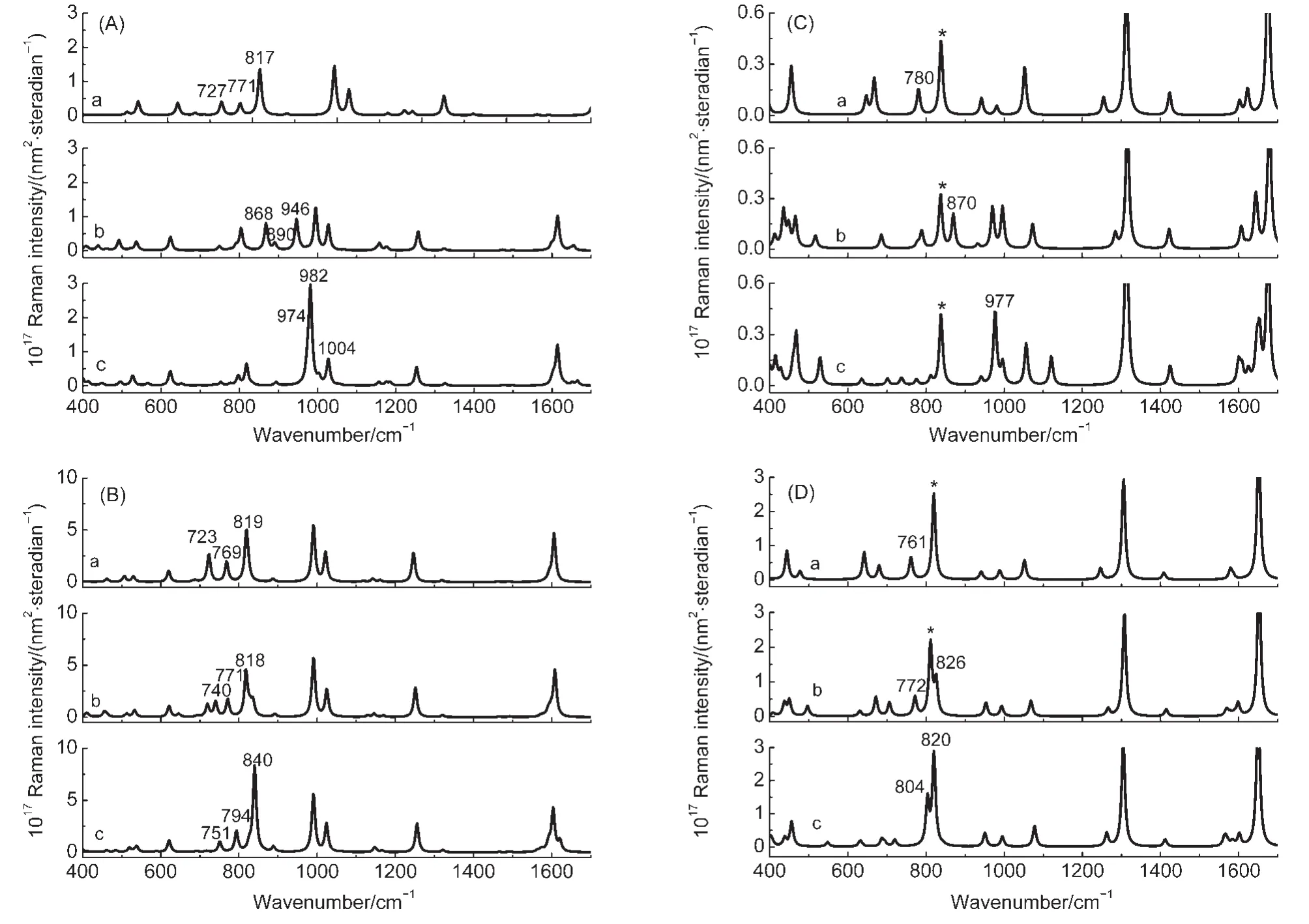
图3 在B3LYP/6-311+G(d,p)水平下计算的苯胺(A,B)和乙烯胺(C,D)与一个(a),三个(b)和五个(c)水分子作用的拉曼光谱图Fig.3 Simulated Raman spectra of aniline(A,B)and vinylamine(C,D)interacting with one(a),three(b),and five(c)water molecules calculated at the B3LYP/6-311+G(d,p)level
图3(B)和3(D)是在溶剂化下苯胺和乙烯胺与水簇复合体系的计算拉曼光谱.考虑溶剂化后,计算拉曼光谱显著不同于在气相条件下计算的拉曼光谱.虽然ωNH2振动谱峰的频率蓝移减小,谱峰强度增加,但计算得到拉曼谱的线形随水簇大小的变化明显减小.对于苯胺分子,氢键作用使ωNH2频率位移至818或840 cm-1.在500-1400 cm-1区间,该拉曼谱峰是较强的拉曼谱峰之一.这与实验测得的苯胺液体和晶体以及苯胺的饱和水溶液的拉曼光谱都非常相似.39-42这是因为三者的氢键作用相近.43但前人将该谱峰指认为苯胺的环呼吸振动44或与氨基有关的其它振动.41依据计算结果,我们认为在苯胺液体中分子间氢键可能导致其ωNH2频率蓝移至820 cm-1,其与苯胺环呼吸振动耦合.在图3(B)中,高波数区有三个相对较强的谱峰,它们依次归属为苯环的三角畸变、苯环上C―H面内弯曲和C―N伸缩振动.它们的振动谱峰在频率和强度上均不随水分子数目变化.图3(D)是在考虑溶剂化后乙烯胺分子的计算拉曼光谱.在溶剂化后,在不同的水簇中其ωNH2频率蓝移程度减小,强度显著增加.如表2所示,在乙烯胺与五个水分子作用时,两种面外弯曲振动发生耦合,且在溶剂化条件下,面外弯曲振动模受水簇大小的影响很小,谱图趋于一致.
上述结果表明,无论苯胺还是乙烯胺,与水簇作用后,氨基面外弯曲振动的频率与甲胺的非常接近.这说明分子之间的弱氢键作用导致苯胺和乙烯胺中氨基的杂化性质与甲胺的接近.为了进一步说明这种振动模对弱相互作用的敏感性,下面我们研究了这两个分子与银簇的吸附作用.
3.3 分子与银簇的作用对氨基面外弯曲振动模的影响
表3给出乙烯胺与四个不同的银簇作用的结构参数和成键能.乙烯胺与银簇作用的成键能在15-59 kJ·mol-1之间,属于弱的化学吸附甚至物理吸附.无论是以氨基还是亚甲基与Ag4作用时,其成键能都是最大的.Ag4簇的这种特殊成键作用主要是它能提供合适的未占据轨道,与氨基氮原子孤对形成配位键.Zhao等18在研究苯胺与银簇的作用中发现,氨基氮上的孤对轨道与Ag4簇的成键最强.Metiu等45,46研究丙烯和DNA碱基与不同银簇作用时,也发现不同配体与Ag4簇作用成键能最大,复合物体系更为稳定.因此,我们主要用Ag4作为表面吸附活性位来模拟分子以氨基吸附在银表面的情况.如图4所示,从成键轨道的电子云图可以看出,当乙烯胺分别以亚甲基和氨基与Ag4簇作用时,分子是以π轨道或者氨基的孤对轨道与Ag4簇的未成键s轨道作用的.
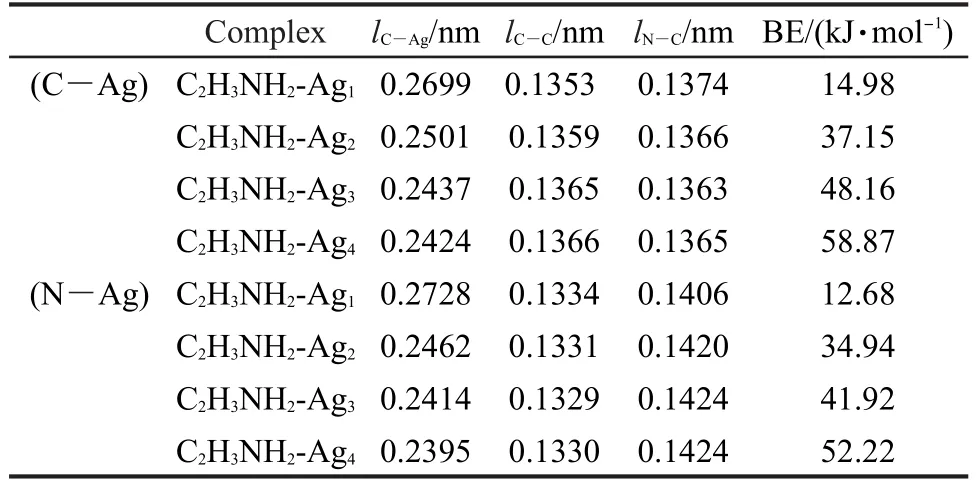
表3 B3LYP/6-311G(d,p)/LANL2DZ(Ag)水平下计算的C2H3NH2-Agn(n=1,2,3,4)复合物的结构参数和成键能Table 3 Structural parameters and bonding energies(BE)for C2H3NH2-Agn(n=1,2,3,4)complexes calculated at the B3LYP/6-311G(d,p)/LANL2DZ(Ag)level
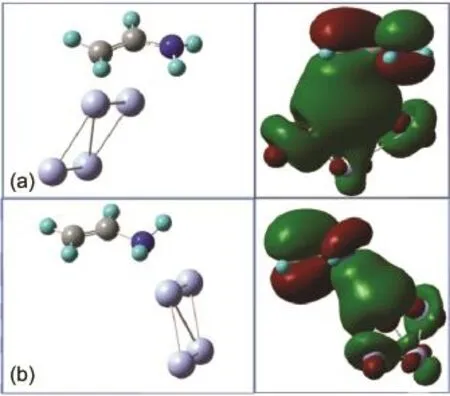
图4 乙烯胺分子分别以亚甲基(a)和氨基(b)与Ag4作用的优化结构及成键轨道图Fig.4 Optimized structures and the plots of bonding orbitals for vinylamine interacting with a silver clusterAg4 via methylene group(a)and amino group(b)
乙烯胺分子同时含有碳碳双键和氨基孤对电子,在自由分子中亚甲基和氨基的面外弯曲振动的频率分别为813和544 cm-1.它在银表面存在两种可能吸附构型,如图4所示,(i)分子以氨基上氮的孤对轨道与银簇作用和(ii)分子以π轨道与银簇作用.我们的计算结果(如表4所示)表明当分子以氨基与银簇作用时,会使分子上氨基与碳碳双键之间的p-π共轭效应减弱.一方面,C―N键长增加,电荷从氮上转移到银簇加强了乙烯胺与银之间的相互作用,此时氨基的翻转角从自由分子中的37.1°增大到48.5°,氨基氮原子的sp3杂化性质增加.这导致ωNH2振动的频率升高到901 cm-1,与自由分子中该振动模的拉曼强度比较,吸附态的拉曼强度增大约15倍.另一方面,这一作用也使C=C键长显著缩短,导致π键的定域性质增强,因此ωCH2振动的频率发生蓝移.当分子以π键与银簇作用时,C=C键长变长,导致ωCH2振动频率降低了5 cm-1.此时靠近氨基碳上的p轨道倾向于与氨基的孤对轨道作用,这使得C―N键长缩短.氨基的面外弯曲振动和摇摆振动耦合并转变为两个NH的面外弯曲振动.
苯胺分子与银簇作用后,氨基的翻转角由自由分子的39.2°增大到50.6°,此时氮原子上的孤对电子与苯环的共轭效应减弱,导致C―N键长伸长,而氨基氮原子的sp3杂化性质增加,因此ωNH2的频率升高到934 cm-1,拉曼强度增大约15倍.比较乙烯胺和苯胺分子以氨基与银簇作用,发现二者的氨基面外弯曲振动模的频率蓝移的程度和拉曼强度增加的倍数非常接近.同时,结果表明弱相互作用对这种p-π共轭体系的氨基面外弯曲振动的影响均较大.

表4 B3LYP/6-311G(d,p)/LANL2DZ(Ag)水平下计算乙烯胺和苯胺与Ag4复合物的翻转角(θ)、C―C键长(l)等结构参数、面外弯曲振动模的频率(ωi)和拉曼强度(Si)Table 4 Inversion angle(θ),structural parameters(l),vibrational frequencies(ωi),and Raman intensities(Si)of wagging modes for C2H3NH2,C6H5NH2and their complexes withAg4calculated at the B3LYP/6-311+G(d,p)/LANL2DZ level
4 结论
通过密度泛函理论计算,分析了苯胺和乙烯胺与水簇的氢键作用以及它们和银簇的弱吸附作用.我们发现这两类弱的相互作用显著影响分子内p-π共轭效应,从而导致分子内氨基面外弯曲振动的拉曼光谱发生显著变化,如在气相中,弱的作用导致拉曼谱峰发生显著蓝移.分子以氨基与水簇或金属簇作用后,氨基氮的sp3杂化性质增强,氨基的翻转角增大导致p-π共轭作用变化.这种弱的作用也导致氨基面外弯曲振动模的拉曼强度增大约15倍.最后,理论研究结果也建议含这类氨基分子的拉曼信号可能用作为一种分子探针,通过分析拉曼信号,研究分子与表面或环境分子的成键性质,推断分子的成键或吸附状态.
(1) Krueger,P.Canadian Journal of Chemistry 1963,41,363.doi:10.1139/v63-053
(2) Xie,M.,X.;Liu,Y.Spectrochimica Acta Part A:Molecular and Biomolecular Spectroscopy 2002,58,2817.doi:10.1016/S1386-1425(02)00072-0
(3) Solca,N.;Dopfer,O.The European Physical Journal D-Atomic,Molecular,Optical and Plasma Physics 2002,20,469.
(4) Szatylowicz,H.;Krygowski,T.M.;Hobza,P.The Journal of Physical Chemistry A 2007,111,170.doi:10.1021/jp065336v
(5)Wolff,H.;Mathias,D.The Journal of Physical Chemistry 1973,77,2081.doi:10.1021/j100636a010
(6) Nakanaga,T.;Ito,F.;Miyawaki,J.;Sugawara,K.;Takeo,H.Chemical Physics Letters 1996,261,414.doi:10.1016/0009-2614(96)00994-3
(7) Nakanaga,T.;Kawamata,K.;Ito,F.Chemical Physics Letters 1997,279,309.doi:10.1016/S0009-2614(97)01059-2
(8) Nakanaga,T.;Sugawara,K.;Kawamata,K.;Ito,F.Chemical Physics Letters 1997,267,491.doi:10.1016/S0009-2614(97)00127-9
(9) Schmid,R.;Chowdhury,P.;Miyawaki,J.;Ito,F.;Sugawara,K.;Nakanaga,T.;Takeo,H.;Jones,H.Chemical Physics 1997,218,291.doi:10.1016/S0301-0104(97)00072-4
(10) Fedotova,M.V.;Kruchinin,S.E.Journal of Molecular Liquids 2013,179,27.doi:10.1016/j.molliq.2012.11.031
(11) Plugatyr,A.;Svishchev,I.M.The Journal of Chemical Physics 2009,130,114509.doi:10.1063/1.3096672
(12) Li,D.W.;Qu,L.L.;Zhai,W.L.;Xue,J.Q.;Fossey,J.S.;Long,Y.T.Environmental Science&Technology 2011,45,4046.doi:10.1021/es104155r
(13) Perry,D.A.;Cordova,J.S.;Schiefer,E.M.;Chen,T.Y.;Razer,T.M.;Biris,A.S.The Journal of Physical Chemistry C 2012,116,4584.doi:10.1021/jp208489w
(14)Wu,D.Y.;Liu,X.M.;Huang,Y.F.;Ren,B.;Xu,X.;Tian,Z.Q.The Journal of Physical Chemistry C 2009,113,18212.doi:10.1021/jp9050929
(15)Tian,Z.Q.;Lei,L.C.;Jing,X.B.Acta Phys.-Chim.Sin.1988,4,458.[田中群,雷良才,景遐斌.物理化学学报,1988,4,458.]doi:10.3866/PKU.WHXB19880504
(16) Park,H.;Lee,S.B.;Kim,K.;Kim,M.S.The Journal of Physical Chemistry 1990,94,7576.
(17) Park,S.H.;Kim,K.;Kim,M.S.Journal of Molecular Structure 1993,301,57.doi:10.1016/0022-2860(93)80231-J
(18)Zhao,L.B.;Huang,R.;Bai,M.X.;Wu,D.Y.;Tian,Z.Q.The Journal of Physical Chemistry C 2011,115,4174.doi:10.1021/jp1117135
(19) Hamada,Y.;Sato,N.;Tsuboi,M.Journal of Molecular Spectroscopy 1987,124,172.doi:10.1016/0022-2852(87)90130-5
(20) McNaughton,D.;Evans,C.J.Journal of Molecular Spectroscopy 1999,196,274.doi:10.1006/jmsp.1999.7861
(21) Brown,R.;Godfrey,P.;Kleibomer,B.;Pierlot,A.;McNaughton,D.Journal of Molecular Spectroscopy 1990,142,195.doi:10.1016/0022-2852(90)90177-R
(22) Marenich,A.V.;Cramer,C.J.;Truhlar,D.G.The Journal of Physical Chemistry B 2009,113,6378.doi:10.1021/jp810292n
(23)Wu,D.Y.;Hayashi,M.;Shiu,Y.J.;Liang,K.K.;Chang,C.H.;Yeh,Y.L.;Lin,S.H.The Journal of Physical Chemistry A 2003,107,9658.doi:10.1021/jp034951l
(24) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09,RevisionA.01;Gaussian Inc.:Wallingford,CT,2009.
(25) McLean,A.;Chandler,G.The Journal of Chemical Physics 1980,72,5639.doi:10.1063/1.438980
(26) Krishnan,R.;Binkley,J.S.;Seeger,R.;Pople,J.A.The Journal of Chemical Physics 1980,72,650.doi:10.1063/1.438955
(27) Wadt,W.R.;Hay,P.J.The Journal of Chemical Physics 1985,82,284.doi:10.1063/1.448800
(28) Hay,P.J.;Wadt,W.R.The Journal of Chemical Physics 1985,82,299.doi:10.1063/1.448975
(29) Schultz,G.;Portalone,G.;Ramondo,F.;Domenicano,A.;Hargittai,I.Structural Chemistry 1996,7,59.doi:10.1007/BF02275450
(30) Boys,S.;Bernardi,F.Molecular Physics 1970,19,553.doi:10.1080/00268977000101561
(31) Reed,A.E.;Curtiss,L.A.;Weinhold,F.Chemical Reviews 1988,88,899.doi:10.1021/cr00088a005
(32) Carpenter,J.;Weinhold,F.Journal of Molecular Structure:Theochem 1988,169,41.doi:10.1016/0166-1280(88)80248-3
(33) Liang,X.J.;Cui,L.;Wu,D.Y.;Tian,Z.Q.Acta Phys.-Chim.Sin.2009,25,1605 [梁晓静,崔 丽,吴德印,田中群.物理化学学报,2009,25,1605.]doi:10.3866/PKU.WHXB20090808
(34) Ziegler,L.;Hudson,B.The Journal of Physical Chemistry 1984,88,1110.doi:10.1021/j150650a016
(35)Tsuboi,M.;Hirakawa,A.Y.;Ino,T.;Sasaki,T.;Tamagake,K.The Journal of Chemical Physics 1964,41,2721.doi:10.1063/1.1726344
(36) Evans,J.Spectrochimica Acta 1960,16,428.doi:10.1016/0371-1951(60)80037-9
(37) Larsen,N.;Hansen,E.;Nicolaisen,F.Chemical Physics Letters 1976,43,584.doi:10.1016/0009-2614(76)80629-X
(38) Rauhut,G.;Pulay,P.The Journal of Physical Chemistry 1995,99,3093.doi:10.1021/j100010a019
(39) Shindo,H.J.Chem.Soc.Faraday Trans.1 1986,82,45.doi:10.1039/f19868200045
(40) Holze,R.Journal of Electroanalytical Chemistry and Interfacial Electrochemistry 1988,250,143.doi:10.1016/0022-0728(88)80199-2
(41) Tripathi,G.The Journal of Chemical Physics 1980,73,5521.doi:10.1063/1.440072
(42)Qi,Y.;Hu,Y.;Xie,M.;Xing,D.;Gu,H.Journal of Raman Spectroscopy 2011,42,1287.doi:10.1002/jrs.v42.6
(43) Funnell,N.P.;Dawson,A.;Marshall,W.G.;Parsons,S.CrystEngComm 2013,15,1047.
(44)Wojciechowski,P.M.;Zierkiewicz,W.;Michalska,D.;Hobza,P.The Journal of Chemical Physics 2003,118,10900.doi:10.1063/1.1574788
(45) Chrétien,S.;Gordon,M.S.;Metiu,H.The Journal of Chemical Physics 2004,121,9925.doi:10.1063/1.1809600
(46) Soto-Verdugo,V.;Metiu,H.;Gwinn,E.The Journal of Chemical Physics 2010,132,195102.doi:10.1063/1.3419930
猜你喜欢
探测与控制学报(2023年4期)2023-09-12 07:26:12
能源化工(2021年6期)2021-12-30 15:41:26
分析科学学报(2021年3期)2021-07-14 01:51:16
色谱(2021年6期)2021-05-06 02:18:56
科技资讯(2020年12期)2020-06-03 04:44:20
合成化学(2015年9期)2016-01-17 08:57:20
现代检验医学杂志(2014年3期)2014-02-02 02:42:29
郑州大学学报(理学版)(2013年3期)2013-03-11 20:30:38
食品科学(2013年24期)2013-03-11 18:30:50
化学分析计量(2013年1期)2013-03-11 16:37:15
