
PVB改性聚氨酯材料及其性能的研究
2013-05-22 01:45:32邓新华韩宇洋岳海生
中国塑料 2013年9期
肖 勇,邓新华,孙 元,韩宇洋,岳海生
(天津工业大学材料科学与工程学院,天津 300387)
0 前言
PU是分子链中含有氨酯键(—NH—COO—)的一类聚合物的统称[1]。由于其具有良好的耐磨性、耐腐蚀性及高弹性等优点[2],在金属防腐、汽车涂装、地板漆、织物涂层等方面得到了广泛的应用。
然而PU在热稳定性、抗形变能力以及环保等方面的性能还不够理想,限制了其在很多领域的进一步应用,本项目组曾采用端胺基非异氰酸酯基PU预聚体与聚醚型聚氨酯预聚体制备了嵌段共聚改性聚醚型PU膜,使PU的热稳定性、抗形变能力有所提高,并减少了残留异氰酸酯基的含量。
在PVB改性PU方面已有一些人进行了尝试,谭正德等[3]以聚酯二元醇、TDI和三羟甲基丙烷为原料,对PVB进行改性,合成出了单组分的三元接枝共聚水性PVB改性PU胶黏剂,赵梓年等[4]利用浸没沉淀相转化法制备了PU/PVB/SiO2共混杂化膜,然而对于PVB用于PU涂层的改性却鲜有人进行研究,本实验室在PU涂层应用的过程中发现PU涂层与基布的结合牢度是限制PU涂层应用的一个重要因素。考虑到PVB大分子上具有较多的可以与异氰酸根反应的活性羟基,PVB与PU预聚体进行扩链反应,可以获得分子链上具有较多仲羟基的PU树脂,该树脂与一些含有极性基团的聚合物材料表面具有较好的相容性和黏结性,可以获得较高剥离强度的复合材料。本文通过调整封闭剂[5]、成膜温度、固化时间、PVB相对分子质量以及配比值等工艺条件制备出新型PU膜,并且对该膜的结构和性能进行了测试和分析。
1 实验部分
1.1 主要原料
PVB1,05-277,工业级,相对分子质量45000,营口天元化工研究所股份有限公司;
PVB2,07-118,工业级,相对分子质量65000,营口天元化工研究所股份有限公司;
PVB3,06-279,工业级,相对分子质量为110000,营口天元化工研究所股份有限公司;
甲苯二异氰酸酯,工业级,深圳鑫隆化工销售有限公司;
聚四氢呋喃醚二醇,工业级,上海市华炜化工有限公司;
甲乙酮肟,分析纯,广州中兰鼎辉材料科技有限公司;
正己烷,分析纯,常州市耕耘化工有限公司;
聚酰胺纤维基布,绍兴县轻纺城千达针织品行。
1.2 主要设备及仪器
电子万能力学试验机,DSS-500,日本岛津公司;
傅里叶红外光谱仪(FTIR),WQF-310,德国布鲁克公司;
热重分析仪(TG),STA 409PC,德国耐驰公司;
动态力学分析仪(DMA),DMA 242,德国耐驰公司;
智能电位滴定仪,808Tritrando,瑞士Metrohm公司。
1.3 样品制备
聚醚型PU预聚体的合成:将150g聚四氢呋喃二醇加入到装有搅拌器、温度计和氮气保护的三颈瓶中,控制在120℃脱水2.5h,降温至80℃,加入55gTDI,预聚2h,制得聚醚型PU预聚体[6],然后用正己烷萃取预聚体中未反应的游离TDI,最后按参考文献[5]中方法用甲乙酮肟对预聚体进行封端处理;
将PVB溶解在丁酮和甲苯的混合溶剂中制成溶液,再将预聚体溶解在此混合溶剂中制成预聚体溶液,按设计的基团比将上述2种溶液按一定比例混合,然后用丁酮甲苯的混合溶剂调整上述混合溶液的黏度制成刮膜胶;用刮刀在转移纸上涂膜,最后将膜在烘箱中加热固化3h,即得到PU膜;采用自制的涂膜机将上述胶液涂在转移纸上,固化后与聚酰胺纤维基布复合,24h后进行剥离强度测试;实验中所用来对比的普通PU均为预聚体经异弗尔酮二胺固化制得,对比试样均在上述步骤且同等条件下进行制备。
1.4 性能测试与结构表征
采用智能电位滴定仪测定正己烷萃取液及PU预聚体的NCO含量,测定方法为二正丁胺法:取约3g的PU预聚体于锥形瓶中,加入15mL甲苯溶液和5mL二正丁胺溶液,静置15min,用移液管加入50mL乙醇,用0.5mol/L的 HCL-C2H5OH 溶液滴定;
FTIR分析:测试范围为650~4000cm-1;
按照GB/T 3923.1—1997进行拉伸性能测试,在室温下测试宽为20mm薄膜的力学性能,拉伸速率为50mm/min,夹持间距为25mm;
剥离强度测试按照GB/T 2791—1995进行测试,试样规格为150mm×25mm×2mm;
TG分析:将样品置于TG分析仪的铝坩锅中进行测试,在氮气氛围保护下进行,从室温缓慢升温至700℃,升温速率为10℃/min;
DMA分析:采用多频双悬臂模式,升温速率为10℃/min,频率为5Hz,温度范围为:-100~200℃。
2 结果与讨论
2.1 膜的FTIR分析
如图1所示,PU预聚体在3300cm-1处有—NH伸缩振动峰,2983cm-1处为C—H的非对称伸缩振动峰;2873cm-1为C—H的对称伸缩振动峰;2277cm-1处有一个—NCO的不对称伸缩振动强吸收峰,1730cm-1处有氨基甲酸酯羰基C O的伸缩振动吸收峰。烷基胺类C—N的伸缩振动峰出现在1121cm-1处,1230cm-1处的吸收峰的出现是由于芳烃C—H变形振动。

图1 样品的FTIR谱图Fig.1 FTIR spectra of the samples
PVB在3441cm-1附近出现了很强的—OH伸缩振动的吸收峰,在2961cm-1处有C—H的伸缩振动峰,1628cm-1为酮类羰基C O的吸收峰,1380cm-1处的吸收峰为—OH的变形振动。此外,在1140cm-1附近还出现了C—O—C的吸收峰。
PVB改性PU的FTIR谱图中除了出现了PU预聚体和PVB的特征峰外,还在1729cm-1处明显出现了—OH同—NCO基团反应生成的—NHCO的吸收峰,且相应的—OH峰减弱,无—NCO的吸收峰,证明预聚体中的—NCO确与PVB中的—OH完全发生了反应。
2.2 PVB相对分子质量及配比对膜力学性能的影响
依照参考文献[6]可知,最佳成膜温度为140℃。在140℃下固化成膜,PVB相对分子质量及配比值对膜力学性能的影响如表1~表3所示。

表1 PVB相对分子质量及配比值对膜断裂强度的影响 MPaTab.1 Influence of PVB molecular weight and the ratio on the fracture strength of the membrane MPa

表2 PVB相对分子质量及配比值对膜断裂伸长率的影响 %Tab.2 Influence of PVB molecular weight and the ratio on the elongation at break of the membrane %

表3 PVB相对分子质量及配比值对膜剥离强度的影响 N/2.5cmTab.3 Influence of PVB molecular weight and the ratio on the peeling strength of the membrane N/2.5cm
由表1~表3可以看出,新型PU膜断裂强度和与聚酰胺纤维基布复合时的剥离强度较普通PU膜有了一定程度的提高,而断裂伸长率则有所下降。这主要是由于新型扩链剂PVB不仅使PU大分子相对分子质量增加,而且在大分子之间形成更多的交联点,使链段移动更为困难,因此材料的拉伸强度有所增加,同时断裂伸长率有所下降。聚酰胺纤维表面大量的酰胺键与新型PU内未反应的羟基相互作用增加了2个界面之间的黏着力,使剥离强度增大。PVB树脂的相对分子质量增大有利于提高膜的断裂强度,且其断裂伸长率也相应地降低。这是由于PVB相对分子质量的增加导致PVB分子链之间的相互作用力增大,因此薄膜的断裂需要更大的外力。而在PVB与PU分子之间形成扩链结构后,大分子之间不易发生相对滑移,故薄膜的断裂伸长率随之降低。随着n(NCO)/n(OH)值的增加,断裂强度与断裂伸长率均是先增加后降低,断裂强度在n(NCO)/n(OH)=1∶1.2处达到最大,而断裂伸长率在n(NCO)/n(OH)=1∶1时最大。理论上,2种官能团间的加成反应,无论何种官能团过量,未反应的官能团会作为分子的端基存在,使大分子的相对分子质量降低,最终导致材料的力学性能受到影响;但是由于PVB中的部分羟基反应活性不高[7],所以PVB略过量增加了羟基与异氰酸基的反应机会,而且基团内部多余的羟基可能与氨基甲酸酯基团形成氢键,因此断裂强度在n(NCO)/n(OH)=1∶1.2处达到最大。另外,随着预聚体n(NCO)/n(OH)值的增加,体系中软段含量增加,硬段含量降低,断裂伸长率逐渐增加,但当大于一定值时,由于分子链过大,分子间不易发生相对滑移,因此断裂伸长率会下降。当n(NCO)/n(OH)过小时,材料虽然含有大量的羟基,但由于膜的柔软度急剧下降,导致剥离强度较小;而n(NCO)/n(OH)较大时,与聚酰胺纤维表面酰胺键的作用较弱,剥离强度也不理想,剥离强度在n(NCO)/n(OH)=1∶1.4处达到最大。
2.3 游离TDI对膜力学性能的影响
由于合成预聚体所用的TDI有2,4-TDI和2,6-TDI 2种结构类型,且2,6-TDI反应活性较低,会有部分残留在预聚体中。为了降低材料的毒性,本文采用正己烷萃取预聚体中的游离TDI,通过滴定萃取液中的NCO含量,检验是否达到去除游离TDI的效果。对二次萃取液进行检测的结果,TDI含量为零,说明预聚体中未反应的游离TDI已被完全萃取;对萃取后PU预聚体进行检测,NCO含量有所下降。分别将萃取后和未萃取的PU预聚体均进行封端处理,再分别与PVB3反应制膜,所得膜的力学性能如表4所示。

表4 游离TDI对膜力学性能的影响Tab.4 Influence of free TDI on mechanical properties of the membrane
由表4可知,经过萃取处理不含游离TDI的固化膜断裂强度较大,且断裂伸长率较大。这是因为预聚体中游离的TDI首先与PVB反应,促进体系黏度不断增大,影响大分子中端异氰酸酯基分子与羟基的反应,使大分子的相对分子质量降低,并且固化时间变长;同时游离的TDI也影响了膜中分子结构的规整性,致使微相区分离的不完善,力学性能下降。
2.4 膜的TG分析
由图2可知,普通PU的起始降解温度为260.5℃,PVB改性PU的起始降解温度为289.9℃,PVB改性PU的起始降解温度升高了29.5℃。理论上,材料热失重率为5%时,其性能会发生变化,普通PU、PVB改性PU、PVB热失重率为5%时对应的温度分别为281.1、300.4、327.7℃。样品失重率为10%时,聚合物的结构与性能可能发生较大变化,不能满足基本使用要求,普通PU、PVB改性PU、PVB对应的热失重率为10%时的温度分别为296.6、310.6、357.3℃。PVB改性后的PU热降解温度[8]有了明显的提高,主要是由于一个PVB大分子中含有多个羟基可与NCO基团进行反应,相当于在高聚物内部产生了交联的效果,使得PVB改性后的PU具有更好的热稳定性。而且在反应中羟基过量,因此在新型PU中会存在更多硬段微区,硬段微区分布于软段相中起着物理交联点的作用,可以显著提高PU的力学性能及热稳定性[9]。

图2 样品的TG曲线Fig.2 TG curves of the samples
2.5 膜的DMA分析
从图3可以看出,3条曲线显示出动态储能模量(E′)随温度的变化趋势是一致的。很显然,PVB改性后的PU在玻璃态E′非常高,并且随温度的升高降低较慢,E′是衡量聚合物材料刚性及弹性的一个重要指标[10],说明在该温度区域,PVB改性PU具有较高的刚性和抗变形能力。这主要是由于新型PU内部的分子链之间作用力较大而且存在交联点,链段以及大分子发生运动需要克服更大的作用力。
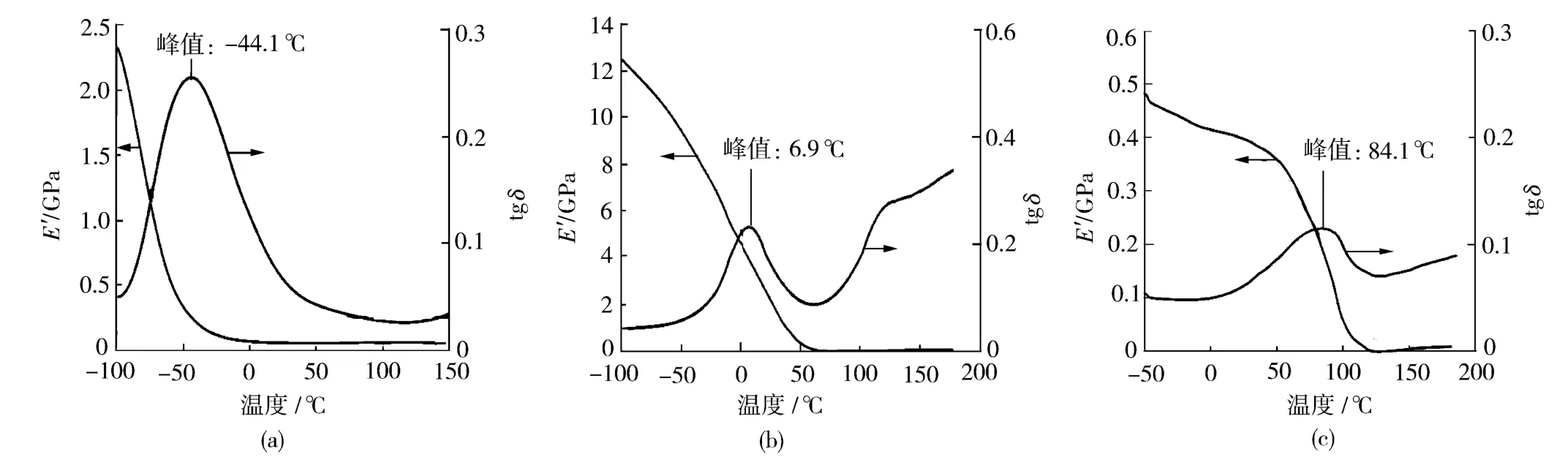
图3 样品的E′和损耗因子(tgδ)随温度的变化Fig.3 Dynamic storage modulus and loss factor of the samples against temperature
由图3可知,在-100~200℃之间普通PU和PVB的损耗因子随温度的变化曲线均出现了一个松弛峰[11],而PVB改性的PU出现了2个松弛峰,第二个松弛峰的范围较宽且接近纯PVB的松弛峰,峰值所对应的温度为玻璃化转变温度(Tg),Tg出现的原因是大分子链段的运动。PVB改性的PU曲线出现2个松弛峰是由于改性PU大分子内存在2种链段:一种为预聚体与PVB反应所生成的链段,对应Tg为6.9℃;另一种为原本存在于PVB分子内的链段,而且由于生成PU产生了交联效果,链段运动更为困难,因此对应的Tg较高。同时链段运动需要更高的温度说明新型PU具有更好的热稳定性,这与上述TG测试所得结果一致。
3 结论
(1)采用PVB作为扩链剂制备出了一种新型PU薄膜材料,这种新型PU材料具有更好的热稳定性和抗变形能力,与聚酰胺纤维基布复合得到的材料具有较大的剥离强度;
(2)当PVB的相对分子质量为110000,n(NCO)/n(OH)=1∶1.2时膜力学性能最佳;
(3)采用萃取过的预聚体制膜,消除了游离TDI对膜性能的影响,使膜的力学性能得到提高。
[1]山西省化工研究所.聚氨酯弹性体手册[M].北京:化学工业出版社,2001:6-7.
[2]WICKS A D,WICKS W Z.Blocked IsocyanatesⅢ:Part A.Mechanisms and Chemistry Progress in Organic Coatings[J].Progress in Organic Coatings,1999,36(3):148-172.
[3]谭正德,杨晓宁,姜 灿.改性PVB单组分聚氨酯胶粘剂[J].中国胶粘剂,2007,16(10):33-37.Tan Zhengde,Yang Xiaoning,Jiang Can.Modified PVB One-component Polyurethane Adhesive[J].China Adhesives,2007,16(10):33-37.
[4]赵梓年,程晓艳.PVB对聚氨酯共混杂化膜微孔结构及性能的影响[J].化工新型材料,2008,36(6):44-46.Zhao Zinian,Cheng Xiaoyan.Effect of PVB on the Microstructure and Performance of PU Composite Membrane[J].New Chemical Materials,2008,36(6):44-46.
[5]殷正虎,邓新华,孙 元,等.封闭剂用于非异氰酸酯预聚体扩链制备聚氨酯弹性体材料的研究[J].中国塑料,2012,26(11):28-31.Yin Zhenghu,Deng Xinhua,Sun Yuan,et al.Study on Sealant Used in Polyurethane Elastomer Prepared by Nonisocyanate Prepolymer Chain Extender[J].China Plastics,2012,26(11):28-31.
[6]宋 赫,邓新华,孙 元.端胺基非异氰酸酯预聚体嵌段共聚聚醚型聚氨酯[J].中国塑料,2012,26(2):37-40.Song He,Deng Xinhua,Sun Yuan.Prepolymers of Aminoterminal Non-isocyanate Block Copolymer Polyether-based Polyurethane[J].China Plastics,2012,26(2):37-40.
[7]苑会林,马沛岚,王 靖,等.聚乙烯醇缩丁醛树脂的性能及应用[J].工程塑料应用,2004,32(2):43-46.Yuan Huilin,Ma Peilan,Wang Jing.Property and Application of Polyvinyl Butyral Resin[J].Engineering Plastics Application,2004,32(2):43-46.
[8]Park Sung C S,Schneider N S.Temperature Dependence of Hydrogen Bonding in Toluene Diisocyanate Based Polyurethanes[J].Macromolecules,1977,10(2):452-458.
[9]林 璟,杨秋转,文秀芳,等.耐热型聚氨酯新型材料热稳定性研究进展[J].化工新型材料,2011,39(6):8-11.Lin Jing,Yang Qiuzhuan,Wen Xiufang,et al.Recent Progress on Thermal Stability of Heat-resistant Polyurethane[J].New Chemical Materials,2011,39(6):8-11.
[10]梁基照.无机粒子填充聚合物复合材料的储能模量及其表征[J].华南理工大学学报,2008,36(11):143-146.Liang Jizhao.Storage Modulus and Its Characterization of Inorganic Particulate-filled Polymer Composites[J].Journal of South China Unibersity of Technology,2008,36(11):143-146.
[11]Xiaohui Liu,Qiuju Wu,Lars A Berglund,et al.Polyamide 6-clay Nanocomposites/Polypropylene-grafted-maleic Anhydride Alloys [J].Polym,2001,42(19):8235-8239.
猜你喜欢
合成技术及应用(2022年1期)2023-01-03 07:20:14
合成材料老化与应用(2022年5期)2022-10-25 07:13:32
合成材料老化与应用(2022年4期)2022-08-25 12:00:54
沈阳理工大学学报(2022年1期)2022-06-09 08:42:24
合成纤维工业(2022年2期)2022-05-06 12:03:08
印制电路信息(2021年10期)2021-12-08 06:00:32
浙江大学学报(理学版)(2016年6期)2016-12-15 03:15:03
分析测试学报(2015年3期)2016-01-13 06:18:12
世界橡胶工业(2015年12期)2015-11-19 02:16:45
武汉纺织大学学报(2014年6期)2014-12-27 01:22:00
