
小分子RNA 干扰鞘氨醇激酶1 表达对APP/PS1 小鼠海马神经元的影响
2013-03-11 08:19孙圣刚
中风与神经疾病杂志 2013年10期
张 远,黎 钢,孙圣刚,乔 娴
阿尔茨海默病(Alzheimer’s disease,AD)影响了全球1.5×107万人次,深入了解神经元变性死亡的机制并找到能够延缓或阻断该靶点的药物是AD研究的核心问题。近年来神经鞘脂(sphingolipid)代谢异常与Aβ 的神经毒性作用受到诸多研究者的关注[1~6]。最早研究发现AD 患者脑标本中神经酰胺神经酰胺(ceramide,Cer)含量增增高,He[7]等的研究发现与正常死亡脑组织标本相比较,AD 患者脑标本的额颞区灰质参与神经鞘磷脂(sphingomyelin,SM)、Cer 代谢的酸性神经鞘磷脂酶及神经酰胺酶活性增强,这导致SM 下降、Cer 含量增高、鞘氨醇(sphingosine,Sph)含量增高,但1-磷酸鞘氨醇(sphingosine1-phospate,s1p)含量下降。在此基础上我们构建了sphk1-siRNA 腺病毒载体,并将其通过立体定位注射的方法转染至APP/PS1 双转基因AD模型鼠海马。前期研究发现老年斑沉积增多,行为学损害程度显著恶化[8],转染后APP/PS1 双转基因小鼠海马组织凋亡程度明显增加[9]。在此基础上我们研究了sphk1-siRNA 转染后对小鼠海马DG 区神经元的影响,现将结果汇报如下。
1 材料与方法
1.1 重组腺病毒载体sphk1-siRNA 的构建重组质粒pDC316 sphk1-siRNA 的构建及病毒载体的构建由上海吉凯生物科技有限公司完成,具体如下:两个编码人类sphk1(GenBank:NM_011451)的序列分别作为siRNA 靶序列和作为阴性对照的非定位导向序列。使用NCBI 数据库筛选,以确保siRNA 靶序列只有sphk1 基因为目标序列。这些寡核苷酸退火形成双链并通过T4 DNA 连接酶将pDC316 载体克隆至U6 启动子的下游,并使用BamHⅠ和HindⅢ生成pDC316-sphk1 siRNA 载体。然后将pDC316-sphk1-siRNA 载体转染至大肠杆菌感受态细胞DH5α 中,使转染后的大肠杆菌感受态细胞DH5α 平铺在含有20mg/ml 卡那霉素的LB-琼脂板的表面。通过琼脂糖凝胶电泳PCR 扩增以确定并挑取阳性克隆,然后通过使用3730 DNA 测序仪(ABI、CA、USA)对阳性菌落进行序列分析。转染前24h,HEK 293 细胞被培养在密度为1.5×106个/60mm2的培养板上。根据Lipofectamine2000 的使用说明,将重组腺病毒质粒和其它辅助质粒共转染到HEK 293 细胞。10~15d 后,细胞病变效应(CPE)逐渐明显,贴壁细胞变得圆润和消融。收集上清液及细胞并在37℃/~70℃反复冻融3 次。含有病毒载体的上清液于4℃以7000 转/min 离心5min 后,使用byAdeno-XTM 纯化试剂盒进行纯化,然后保存在-70℃备用。斑点法检测重组病毒的滴度[9]。
1.2 实验动物 根据实验动物管理标准的指导,11 个月年龄的APPsw/PS1(APP/PS1)双转基因小鼠(由南京大学模式动物研究所提供)(n=12)和野生型小鼠组(n=6)饲养在华中科技大学同济医学院动物房。在用12h 光照和12 个黑暗循环的恒温环境中,每笼3 只。动物根据自身需要自由采食食物和水。其中注射sphk1-siRNA 者为siRNA 组,注射生理盐水者为生理盐水组,同型野生型小鼠为阴性对照组(n=6)。
1.3 立体定位注射法 APP/PS1 双转基因小鼠及同型野生型小鼠经10%水合氯醛0.6ml/kg 麻醉后,固定于携带0.5μl 汉密尔顿微量注射器的脑立体定位仪上。头顶部去毛,消毒皮肤,沿正中切口,经双氧水消毒后,充分暴露前囟。根据小鼠脑立位图谱,于前囟后1.9mm、中-外左右各1.4mm、背-腹上下1.5mm 处,向双侧海马缓慢注入1.5μl/位点(1×1011PFU/ml)的sphk1-siRNA 载体、生理盐水及rAd-GFP,每侧注射时间5min。后将针头留在原先的位置60s 使溶液远离针尖并充分扩散,缓慢退针。皮肤切开处用青霉素抗菌,缝合伤口。待小鼠苏醒后送回动物房饲养,密切观察小鼠的变化。
1.4 Western-blot 法 取出海马后,称量剪碎。加入400μl 单去污剂裂解液(含PMSF),充分匀浆。4℃下12000rpm 离心5min。BCA 计算出蛋白浓度,配制10%的分离胶10ml 及4%的浓缩胶5ml,4℃电压12V 转膜过夜;再加入5μl 一抗,室温下在摇床上缓慢摇动1h,再加入PVDF 膜与辣根过氧化物酶抗小鼠二抗(1∶5000)37℃孵育1h;5%脱脂奶粉(含0.05%Tween 20)洗3 次。ECL 化学发光法:在暗室中,按照1∶1 的比例等量混合ECL 化学发光试剂盒中的A、B 两液,混匀后立即滴加于PVDF 膜表面(注意:滤膜的蛋白面应朝上),室温下孵育1~2min,然后用滤纸吸干滤膜表面多余的液体,将滤膜夹入透明塑料袋中,EPSON 彩色图像扫描仪扫描成图像文件并用Kodak Digital Science ID2.0 分析软件分析条带的灰度值,计算同一份标本的PAR-1 和PAR-2 蛋白与actin 蛋白灰度值的比值以对蛋白表达进行半定量分析。
1.5 免疫荧光法显微镜观察 腹腔灌注4%的多聚甲醛及生理盐水后分离脑组织,并将其在4%多聚甲醛中固定,待脑沉底后用冰冻切片机切片,厚度为10~12μm,胶水固定在载玻片上。后将玻片浸入4%多聚甲醛中固定10min,再用0.01 PBS漂洗5min×3 次。然后0.2% Triton X100(20%Triton X100+0.01PBS 100ml)室温孵育30min,以增加细胞的通透性,PBS 清洗5min×3。并用5%胎牛血清(BSA)的0.01mol/L PBS 封闭20min,室温孵育20min。倒掉封闭液,加入一抗 sphk1(羊抗,1∶150),放入湿盒,37℃孵育,1h 后转入4℃过夜,PBS 漂洗5min×3 次后加入罗丹明标记的二抗(兔抗,1∶100),37℃,1h。然后再次加入一抗Abeta(兔抗,1∶200),37℃孵育,1h 后转入4℃过夜,0.01PBS 漂洗5min×3 次并加入Dylight 405 蓝色荧光二抗(羊抗,1∶100),37℃,2h。PBS 漂洗5min×3次后,使用碳酸甘油缓冲液封片,并用激光共聚焦显微镜观察(Olympus、Tokyo、Japan)进行观察。
1.6 电镜超微结构观察 将实验动物经10%水合氯醛6ml/kg 腹腔麻醉后,按常规经左心室升主动脉灌注固定;然后开颅取脑,剥离海马。置入4℃的2.5%电镜专用戊二醛溶液中固定2h;后使用0.1 mol PBS 漂洗2 次,10min/次;再用1%四氧化锇后固定2h;后丙酮梯度脱水,浸透、包埋、固化;并采用半薄切片定位及超薄切片,铀染,铅染,最后行电镜(Hitachi H-7500)观察。
1.7 统计学处理 使用SPSS 13.0 软件(SPSS Inc,USA)进行统计分析,所有数据用均值±标准差(±s)表示。平均值的比较采用t 检验,P<0.05其差异有统计学意义。
2 结果
2.1 sphk1-siRNA 对APP/PS1 双转基因AD模型鼠海马超微结构的影响 转染后1 个月,我们对siRNA 组、生理盐水组、阴性对照组小鼠海马组织行电镜检查。结果如图1 所示:图A 相对于图C来说核质变空和部分神经元胞质变空,但线粒体和内质网整齐。图B 相对于图C 来说可见微丝和微管消失,细胞核核质减少,内质网凌乱,神经元不饱满。由此可见成功转染sphk1-siRNA 后,海马超微结构变性程度加重(见图1)。
2.2 sphk1-siRNA 对APP/PS1 双转基因AD模型鼠海马DG 区新生神经元的影响 为了进一步检测sphk1-siRNA 转染后对小鼠DG 区新生神经元的影响,我们采用激光共聚焦的方法对其进行检测。结果显示:阴性对照组小鼠DG 区可见新生神经元形成,但是生理盐水组及siRNA 组未见明显新生神经元形成。由此可见siRNA 组及生理盐水组小鼠突触神经元再生功能受到抑制(见图2)。
2.3 sphk1-siRNA 对APP/PS1 双转基因AD模型鼠海马组织s1P 受体的影响 由于s1p 通过G蛋白偶联发挥其生物学作用且影响突触后递质的传递,所以我们采用real-time PCR 对小鼠是s1p 受体(s1pr)进行了检测,结果发现siRNA 组s1pr1 表达明显低于其他两组,而s1pr3 表达则明显高于其他两组。
2.4 sphk1-siRNA 对APP/PS1 双转基因AD模型鼠突触后膜蛋白SNAP-25 表达的影响 为了进一步证实sphk1 表达下降后对突触后神经元的影响,我们采用Western-blot 的方法对APP/PS1 突触后膜蛋白SNAP-25 进行了检测,结果显示siRNA 组SNAP-25 表达明显低于其他两组。
3 讨论
阿尔茨海默病是一种最常见的渐进性大脑退行性病变,其发病率随年龄增长逐渐增高。临床上主要表现为进行性认知功能障碍。而其主要的神经病理学以β 淀粉样蛋白(amyloid beta-protein,Aβ)沉积为核心的老年斑、神经元内的神经原纤维缠结和明显的胶质化为特征。迄今其确切的病因和发病机制尚未充分阐明,尚无有效的根治良策。
神经鞘脂(sphingolipid)代谢物包括神经鞘磷脂、神经酰胺、鞘氨醇和1-磷酸鞘氨醇等多种代谢产物,目前研究发现这些磷脂的代谢产物参与细胞增殖与凋亡的调控。Cer 和Sph 是细胞增殖的负调控子,能够抑制细胞生长、促进细胞凋亡;而其下游的代谢物s1p 则刺激细胞生长、抑制细胞凋亡[10]。S1p 既是细胞内信号转导的信使分子,又可以分泌至细胞外并通过细胞表面的s1p 受体发挥生物学效应。细胞内Cer、Sph 和s1p 的水平通过酶促反应维持动态平衡,从而维持细胞的生理功能并决定细胞的生存和凋亡,因此,有人将由Cer、Sph 和s1p 共同构成的动态体系形象地称作“鞘磷脂变阻器”(sphingolipic rheostat)[5]。鞘氨醇激酶是“鞘磷脂变阻器”的关键调节子。sphk1 将Sph 转化成s1p,不仅能够增加促生长、抗凋亡的信号分子s1p 水平,而且能够减少促凋亡的Cer 和Sph 的水平。因此sphk1 是维持细胞内上述物质平衡的重要限速酶,也是细胞增殖及存活的重要调控因子[10]。
突触功能障碍常发生在AD 病理改变的早期且为具有标志性的改变。Aβ 沉积导致网络活动异常兴奋和涉及学习和记忆通路代偿性的抑制反应,这些改变都会促进认知能力下降[11]。研究发现s1p参与少突胶质细胞[12]、中枢神经系统髓鞘细胞[5]的生长和存活以及调节神经元的兴奋性[13]。海马神经元中的s1p 通过s1p 受体来促进神经递质的释放[14],且与s1p 受体偶联的G 蛋白与激活下游信号通路不同效应的分子具有不同的亲和力[15]。S1pr1可以调节神经轴突的形成/延伸,s1pr3 则刚好起着相反的作用[16]。例如,sphk1 的激活和s1p 的形成可以保护中脑神经元免受谷氨酸诱导的神经毒性的影响[17]。多巴胺诱导的谷氨酸盐的释放依赖于sphk1 的激活及s1p 的形成以及相应的s1p 受体的产生,表明s1p 通过其自分泌的作用促进海马神经元中谷氨酸的分泌[15]。
综上所述,s1p 的形成可以通过上调s1p 和它的受体的表达提高突触的活性。我们的研究表明,sphk1-siRNA 转染后神经酰胺减少的同时促进s1p 的产生具有重要意义。同时,Aβ 寡聚体病理性的沉积抑制了突触兴奋性的传递,但也引发了神经回路和网络异常的痫样放电模式[18]。SNAP-25 作为突触后膜SNARE 复合体组成之一,在sphk1-siRNA 转染后表达明显减少,从另一方面说明了突触活性下以及学习和记忆能力显著恶化的原因。
由此可见,sphk1-siRNA 转染后抑制了APP/PS1双转基因小鼠海马DG 区神经元的再生及突触的功能,这进一步丰富并完善了之前的研究,揭示了sphk1基因沉默后APP/PS1 双转基因小鼠行为学障碍加重的原因,同时也为进一步探索sphk1 高表达逆转阿尔茨海默病双转基因模型鼠病理改变提出了新的可能。
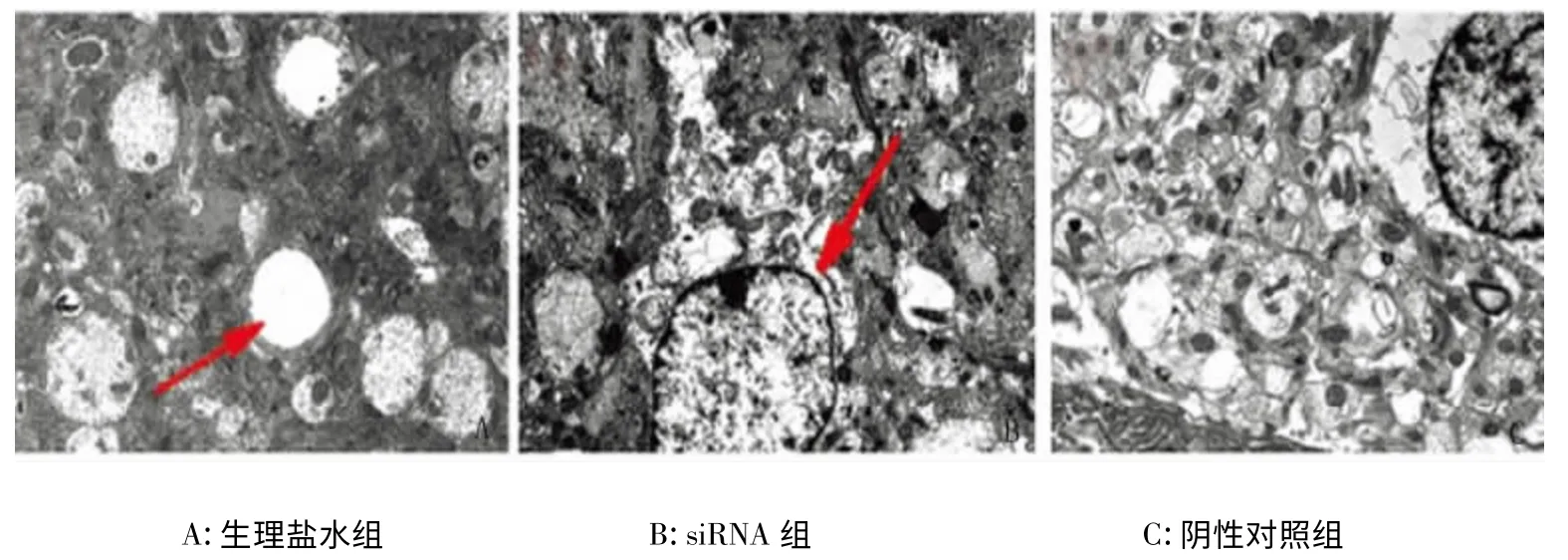
图1 转染Sphk1-siRNA 后海马超微结构的改变
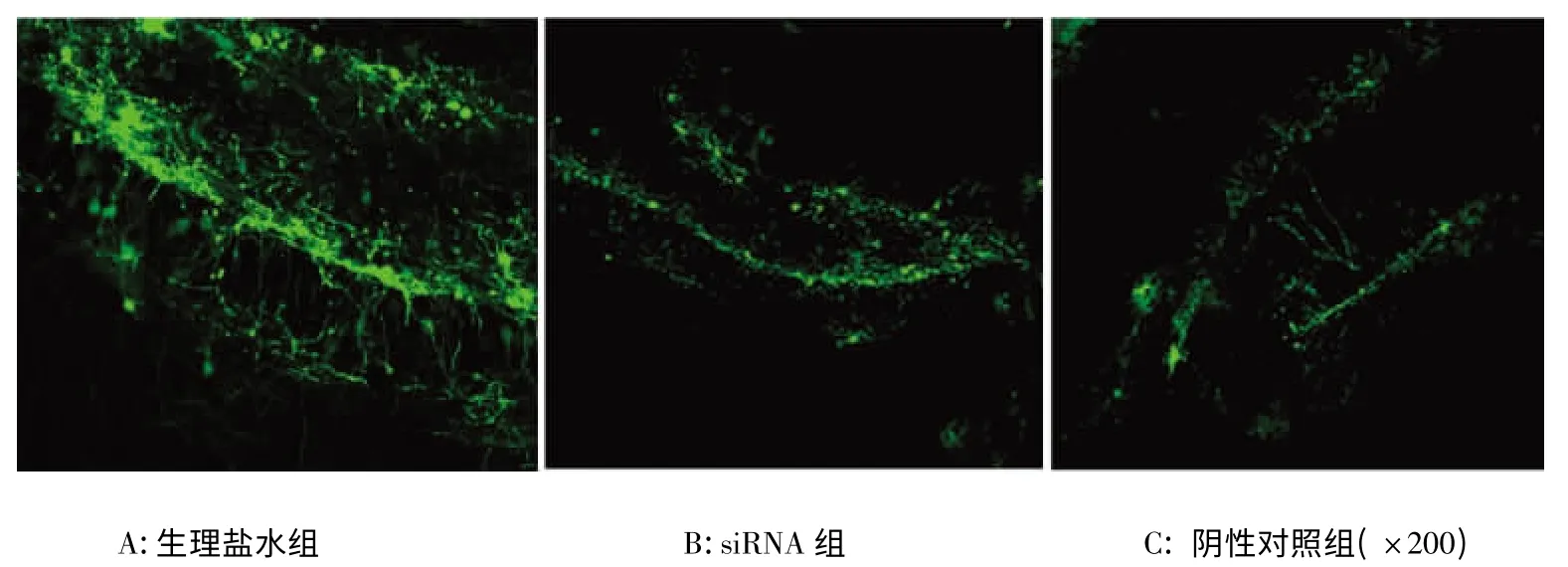
图2 转染Sphk1-siRNA 后海马DG 区神经元的改变
[1]Yankner BA.New clues to Alzheimer’s disease:unraveling the roles of amyloid and tau[J].Nat Med,1996,2850-2852.
[2]Yankner BA,Lu T,Loerch P.The aging brain[J].Annu Rev Pathol,2008,3:41-66.
[3]Hannun YA,Obeid LM.The Ceramide-centric universe of lipid-mediated cell regulation:stress encounters of the lipid kind[J].J Biol Chem,2002,277:25847-25850.
[4]Spiegel S,Milstien S.Sphingosine-1-phosphate:an enigmatic signaling lipid[J].Nat Rev Mol Cell Biol,2003,4(5):397-407.
[5]Singh IN,Hall ED.Multifaceted roles of sphingosinephosphate How does this bioactive sphingolipid fit with acute neurological injury[J].J Neurosci Res,2008,86(7):1419-1433.
[6]Morales A,Lee H,Goni FM,et al.Sphingolipids and cell death[J].Apoptosis,2007,12(5):923-939.
[7]He X,Huang Y,Li B,et al.Deregulation of sphingolipid metabolism in Alzheimer’s disease[J].Neurobiol Aging,2010,31(3):398-408.
[8]Zhang Y,Yu Q,Lai TB,et al.Effects of small interfering RNA targeting sphingosine kinase-1 gene on the animal model of Alzheimer’s disease[J].J Huazhong Univ Sci Technolog Med Sci,2013,33(3):427-432.
[9]张 远,禹 虔,黎 钢,等.小分子RNA 干扰鞘氨醇激酶1 表达对APP/PS1 小鼠海马凋亡的影响[J].神经损伤与功能重建,2013,8(4):239-242.
[10]Okada T,Kajimoto T,Jahangeer S,et al.Sphingosine kinase/sphin-gosine 1-phosphate signalling in central nervous system[J].Cellul Signal,2009,21(2):7-13.
[11]Leonard AS,McNamara JO.Does epileptiform activity contribute to cognitiveimpairment in Alzheimer’s disease[J].Neuron,2007,55(5):677-678.
[12]Yu N,Lariosa-Willingham KD,Lin FF,et al.Characterization of lysophosphatidic acid and sphingosine-1-phosphate-mediated signal transduction in rat cortical oligodendrocytes[J].Glia,2004,45(1):17-27.
[13]Zhang YH,Fehrenbacher JC,Vasko MR,et al.Sphingosine-1-phosphate via activation of a G-protein-coupled receptor(s)enhances the excitability of rat sensory neurons[J].J Neurophysiology,2006,96(3):1042-1052.
[14]Kajimoto T,Okada T,Yu H,et al.Involvement of sphingosine 1-phosphate in glutamate secretion in hippocampal neurons[J].Mol Cell Biol,2007,27(9):3429-3440.
[15]Ishii I,Fukushima N,Ye X,et al.Lysophospholipid receptors:signaling and biology[J].Annu Rev Biochem,2004,73:321-354.
[16]Toman RE,Payne SG,Watterson KR,et al.Differential transactivation of sphingosine-1-phosphate receptors modulates nerve growth factorinduced neurite extension[J].J Cell Biol,2004,166(3):381-392.
[17]Shinpo K,Kikuchi S,Moriwaka F,et al.Protective effects of the TNFceramide pathway against glutamate neurotoxicity on cultured mesencephalic neurons[J].Brain research,1999,819(1):170-173.
[18]Palop JJ,Mucke L.Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease:from synapses toward neural networks[J].Nat Neurosci,2010,13(7):812-818.
猜你喜欢
学与玩(2022年10期)2022-11-23
疯狂英语·初中天地(2022年5期)2022-07-06
今日农业(2022年3期)2022-06-05
疯狂英语·初中版(2022年5期)2022-05-11
中风与神经疾病杂志(2021年9期)2021-11-08
中成药(2017年9期)2017-12-19
中成药(2017年5期)2017-06-13
医学信息(2016年31期)2017-02-27
标记免疫分析与临床(2016年9期)2016-11-21
创新科技(2015年1期)2015-12-24
