
过渡金属M(M=Cu,Ag,Au)对X…Cl(X=F,Cl,Br)卤键相互作用强度的影响
2012-12-11 09:34:26冯大诚
物理化学学报 2012年6期
赵 强 冯大诚
(1淄博职业学院化学工程系,山东淄博255314;2山东大学理论化学研究所,济南250100)
过渡金属M(M=Cu,Ag,Au)对X…Cl(X=F,Cl,Br)卤键相互作用强度的影响
赵 强1,*冯大诚2
(1淄博职业学院化学工程系,山东淄博255314;2山东大学理论化学研究所,济南250100)
采用量子化学方法,通过MCH2X…ClF(M=Cu,Ag,Au;X=F,Cl,Br)和CH3X…ClF两类复合物的对比,探讨了过渡金属对卤键相互作用强度的影响.CH3X…ClF复合物只有卤键相互作用,而优化MCH2X…ClF复合物除了得到一种只含有卤键相互作用的构型外,还得到一种含有过渡金属和Cl原子相互作用的稳定构型.含有过渡金属的复合物稳定性明显增加,Ag取代的复合物稳定性增加最为明显,Cu次之,Au最不明显.X原子最负分子表面静电势(MEP)减小是复合物稳定性增加的根本原因.利用自然键轨道(NBO)及分子中原子(AIM)分析进一步对体系的分子间相互作用进行了探讨.二阶稳定化能与键鞍点处拓扑性质的计算结果与相互作用能符合得很好.
卤键;过渡金属;增强效应;稳定性
1 引言
分子间相互作用在化学的诸多领域引起了人们越来越广泛的关注,氢键相互作用已经被广泛研究.1,2另外,由卤原子(路易斯酸)和中性或带负电的路易斯碱之间形成的相互作用也成了研究的热点.为了强调和传统氢键的相似性,这种以卤原子为电子受体的相互作用被称为卤键.3-9Politzer等6用分子静电势解释了共价卤原子最外层正的静电势区域产生的原因,他们称之为“σ-hole”,并且发现这个区域随着卤原子的半径增大而增大.另外,R―X (X=F,Cl,Br,I)分子的R基团所连原子基团吸电子能力越强,此正静电势区域越大.由于它的强度、选择性和方向性,卤键已经成为设计新功能材料的一个有力工具.10-12
最近,一种特殊的卤键相互作用,C—X…Xʹ—M(M通常是过渡金属元素)被大量报道.13-20与传统卤键C—X…Y(Y是电子给体)不同的是,这类卤键相互作用由有机卤化物C—X和无机卤化物Xʹ—M组成.研究表明,这类复合物的强度比一般卤键复合物大,并且呈现出特殊的物理性质,比如导电性、磁性以及非线性光学性质等.Brammer等15发现,当卤原子作为电子给体时,trans-[PdCl2(Me)(PH3)2]中Cl原子的最负静电势为-221.8 kJ·mol-1,而在CH3Cl中,此值仅为-79.5 kJ·mol-1.Xu等21用量子化学方法研究了PyCl…X(PyCl=NC5H4Cl-4;X=F-, Cl-,Br-)和MPyCl…X(M=Cu+,Zn2+)的卤键相互作用,他们发现,引入的过渡金属元素对卤键相互作用有很大的加强.他们从分子静电势、电子密度差以及轨道组成等几个方面分析了过渡金属元素对卤原子的影响,得出了相似的结论.Bertani等22认为,可以通过改变Xʹ—M中过渡金属的种类,从而改变分子间相互作用的强度,进而达到调控超分子结构的目的.
以上研究结果表明,无论卤原子作为电子给体还是电子受体,过渡金属的引入都会使卤键增强.鉴于含有过渡金属元素的卤键相互作用的理论研究目前还较少,21本文系统研究了MCH2X…ClF(M= Cu,Ag,Au;X=F,Cl,Br)的卤键相互作用.通过与不含有过渡金属的复合物CH3X…ClF进行对比,从几何结构、相互作用能和静电势几个方面探讨了过渡金属对卤键强度的影响.我们还对这些体系进行了自然键轨道(NBO)和分子中的原子(AIM)分析,揭示了卤键增强的化学本质.
2 计算方法
所有单体和复合物的几何结构都用MP2方法进行优化,频率计算在相同的水平下进行,以确保得到的结构是势能面上的最小点.基组的选择如下:非金属原子采用6-311++G(d,p)基组,Cu原子采用Wachter+f基组,23Ag和Au采用赝势基组LANL2DZ.24复合物的能量在相应的计算水平下进行了基组重叠误差(BSSE)校正.25以上的计算都是在Gaussian 03程序26中完成的.NBO计算27利用Gaussian 03中的NBO3.0程序完成.AIM计算在AIMAll程序28中进行,输入的是在Gaussian 03程序中产生的MP2计算水平下的波函数.
3 结果与讨论
3.1 几何结构
首先优化不含有过渡金属元素的CH3X(X=F, Cl,Br)…ClF复合物,只得到了一种稳定结构,如图1所示.这种复合物属于CS点群,CH3X分子中的两个H原子朝向ClF分子中的Cl原子.三种复合物的相互作用能分别为-13.052、-12.801和-12.985 kJ· mol-1,X…Cl之间的距离分别为0.267、0.307和0.316 nm.
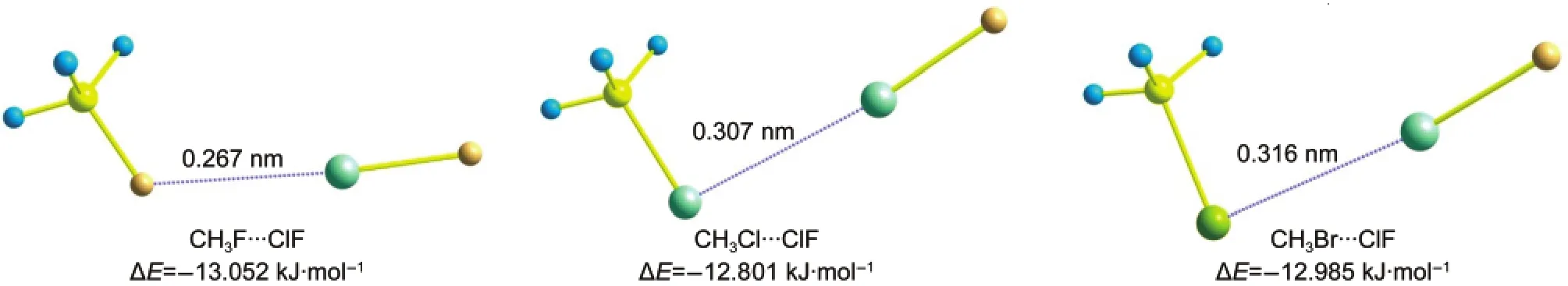
图1 优化得到的CH3X···ClF(X=F,Cl,Br)复合物的几何结构、分子间相互作用距离和相互作用能Fig.1 Optimized geometries,intermolecular interaction distances,and the interaction energies for complexes CH3X···ClF(X=F,Cl,Br)
随后优化了含有过渡金属Cu、Ag和Au的MCH2X…ClF复合物.与不含有过渡金属元素的卤键复合物只有一种能量最小的结构相比,含有过渡金属元素的复合物有两种稳定结构,相应的示意图和定义的几何量在图2中给出.所得到的两种结构同样具有CS点群对称性.其中,structure I的结构与CH3X…ClF类似,即两个H原子朝向Cl原子,而金属原子远离Cl原子.Structure II则与structure I有着明显的不同.第二种结构中除了传统的X…Cl卤键相互作用外,还有过渡金属原子M与Cl原子的M…Cl相互作用.这类分子间相互作用是非常特殊的,因为金属原子和Cl原子所带电荷皆为正,也就是两个带正电荷原子之间的相互作用.Wang等29研究了FBr双分子形成的复合物,发现可以形成FBr…BrF的稳定结构.其中,Br原子都带正电荷,但是正电荷与正电荷的相互作用却能稳定存在,这与本文的M…Cl相互作用类似.值得注意的是,对于含有Cu和Ag的复合物,两种结构都是势能面上的稳定结构,而对于含有Au的复合物来说,只有structure I是稳定结构,structure II有一个虚频,继续优化以后转化为structure I.因此,在后面的讨论中,我们不分析含有Au复合物的第二种结构.
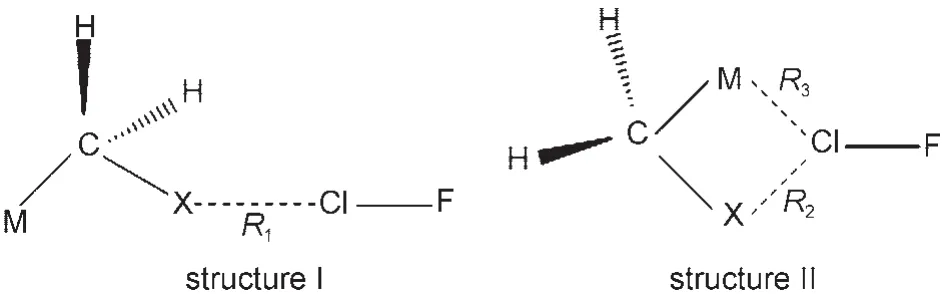
图2 MCH2X···ClF(M=Cu,Ag,Au)复合物的二种几何结构示意图及定义的参数Fig.2 Schematic map for the two structures of the MCH2X···ClF(M=Cu,Ag,Au)complexes and the defined parameters
表1列出了MCH2X…ClF复合物中相应的几何参数,其中,R1和R2分别是structure I和structure II中X…Cl的距离,R3是structure II中M…Cl的距离.从表1中可以看到,相对于CH3X…ClF复合物,在MCH2X…ClF复合物中,X…Cl的距离都缩短了.例如,CH3Cl…ClF中Cl…Cl的距离是0.307 nm,当一个H原子被金属原子取代后,所形成的structure I复合物CuCH2Cl…ClF、AgCH2Cl…ClF和AuCH2Cl… ClF中Cl…Cl的距离分别为0.297、0.293和0.303 nm.对于structure II复合物,这个距离缩小得更为显著.与氢键类似,同类卤键的强度也与原子间距离有着非常好的线性关系,一般来说,距离越小,强度越大.这说明,当CH3X中的一个H原子被过渡金属原子取代后,卤键的强度变大了.再来看过渡金属种类对卤键强度的影响.对于X原子相同的复合物来说,Ag取代的结构R2相比R1减小的幅度最大,其次是Cu,Au的最小.例如,CH3F…ClF复合物中F…Cl的距离是0.267 nm,AgCH2F…ClF(I)中F…Cl的距离是0.254 nm,减小了0.013 nm,CuCH2F…ClF(I)中F…Cl的距离是0.257 nm,减小了0.010 nm,而AuCH2F…ClF(I)中F…Cl的距离是0.263 nm,只减小了0.004 nm.从表1中还可以看出,对于同一类复合物,R2要比R1小,也就是说,structure II中的卤键要比structure I中的强.这说明,structure II复合物中M…Cl的分子间相互作用使卤键X…Cl得到了加强.
表2列出了MCH2X…ClF复合物中C—X键与C—M键的键长在形成复合物前后的变化值.在structure I复合物中,C—X键的键长都变大了,但是C—M键的变化方向却不确定;而在structure II复合物中,C—X键和C—M键都伸长了.这与上文的结果是一致的.当过渡金属原子取代H原子后,structure I复合物的分子间距离减小,但是此时金属原子并没有直接参与分子间相互作用.在structure II复合物中,C—X键和C—M键同时参与分子间相互作用,加上卤键强度的增加,所以变化趋势相同.另外,Δr(C—X)(II)大于Δr(C—X)(I),这与上文“M…Cl的分子间相互作用使卤键得到了加强”的观点也是一致的.

表1 优化得到的MCH2X···ClF复合物的几何参数Table 1 Optimized geometric parameters for the MCH2X···ClF complexes
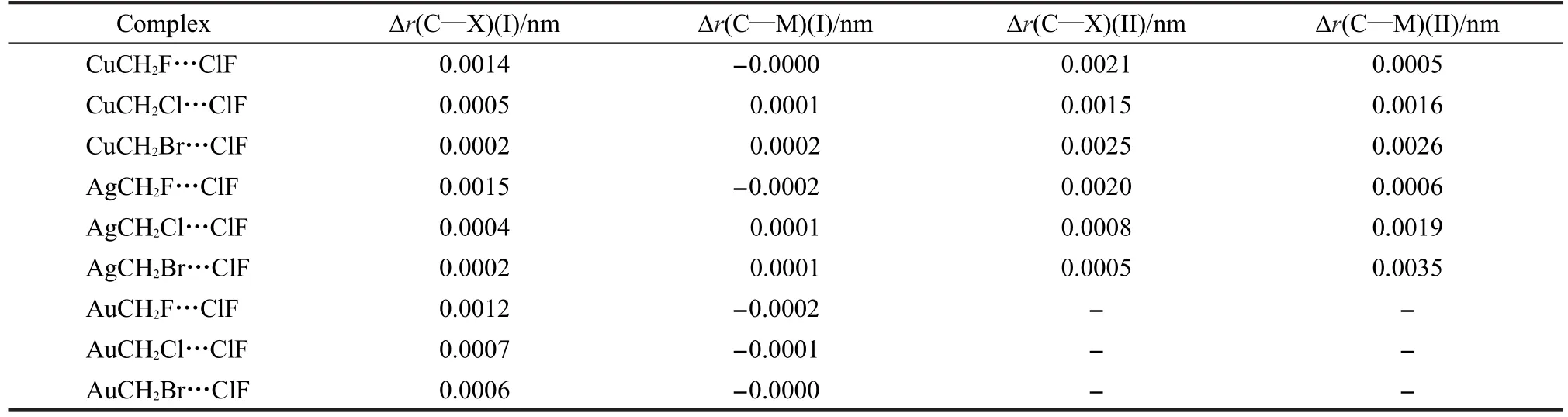
表2 MCH2X···ClF复合物中C―X键与C―M键在形成复合物前后的变化值Table 2 Variation of the bond lengths of C―X and C―M bonds before and after the formation of MCH2X···ClF complexes
3.2 相互作用能
MCH2X与ClF形成的复合物的BSSE校正的相互作用能列在表3中.其中,ΔEI是structrue I复合物的相互作用能,ΔEII是structrue II的相互作用能.很明显可以看出,与CH3X…ClF复合物相比, MCH2X…ClF复合物更加稳定.从表3中还可以看出,structrue II复合物要比structrue I复合物更为稳定,这与前文提到过的structrue II中存在两种弱相互作用是一致的.对于X原子相同的structrue I复合物来说,Ag取代的卤键复合物最稳定,其次是Cu, Au取代的复合物稳定性最差.这与前文得到的键长变化的结果是一致的.值得注意的是,以上提到的复合物稳定性顺序与三种过渡金属离子化能从大到小的顺序正好相反(实验上测定的Au离子化能最大,Cu次之,Ag最小30).对于structure II复合物来说,情况有些复杂,AgCH2F…ClF和AgCH2Cl…ClF复合物的稳定性比相应的Cu取代复合物强,但CuCH2Br…ClF的相互作用能却比AgCH2Br…ClF的更负.

表3 CH3X…ClF及两种不同结构的MCH2X…ClF复合物的相互作用能Table 3 Interaction energies of CH3X…ClF and the two types of MCH2X…ClF complexes
Politzer等31指出,与氢键相互作用类似,卤键相互作用也是由静电相互作用驱动的分子间相互作用.他们通过计算相应成键原子上的分子表面静电势(MEP),发现复合物的稳定性与静电势的大小有很好的一致性.本文分别计算了CH3X和MCH2X分子中X原子在0.001 a.u.电子密度等值面上的最负分子表面静电势.此电子密度等值面被Politzer等6证实可以包含90%以上的电子,所以本文也采用这样的等值面进行计算.所得结果示于图3.从图3中可以看出,随着过渡金属取代H原子,卤原子上负静电势都有不同程度的减小,这与前文中的MCH2X…ClF复合物稳定性增加是一致的.由图中还可以看出,AgCH2X分子中X原子的负静电势减小的最多,而AuCH2X分子中X原子的负静电势减小的最少,几乎与CH3X分子一样.这与前文得到的键长和相互作用能数据是一致的,也从另外一个角度解释了为什么AuCH2X与ClF不能形成structure II结构的复合物.由以上两个方面可以看出, CH3X…ClF和MCH2X…ClF的structrue I复合物中的卤键相互作用仍然是由静电相互作用驱动的分子间相互作用.
3.3 自然键轨道分析

图3 MCH2X分子中X原子最负分子表面静电势(MEP)随M变化的关系图Fig.3 Relationship between the types of M and the most negative molecular electrostatic potential(MEP) of X atom in MCH2X
氢键和卤键的强弱还可以用电子给体的孤对电子轨道和电子受体的反键轨道之间的相互作用来描述.27本文采用NBO方法分析了CH3X…ClF和 MCH2X…ClF复合物中的轨道相互作用.分析结果表明,对于CH3X…ClF和MCH2X…ClF中的structure I复合物来说,轨道相互作用主要发生在X的孤对电子轨道(LPX)和F—Cl键的反键轨道σ*(F—Cl)之间.在MCH2X…ClF中的structure II复合物中,除了X的孤对电子轨道(LPX)和F—Cl键的反键轨道σ* (F—Cl)之间存在轨道相互作用外,FCl分子中Cl原子的孤对电子轨道(LPCl)与MCH2X分子中C—M键的反键轨道σ*(C—M)之间也存在相互作用.Wang等29用NBO方法分析了FBr…BrF复合物,发现之所以两个带正电荷的原子存在稳定的相互作用,是Br原子的孤对电子与F—Br键的反键轨道之间存在强烈的相互作用所致,这与structure II复合物中M…Cl相互作用的分析是一致的.
表4列出了这些轨道相互作用的二阶稳定化能(E(2)).从表4中可以看出,相比于CH3X…ClF复合物来说,MCH2X…ClF复合物中E(2)[LPX→σ*(F—Cl)]的值都增大了.例如,CH3F…ClF复合物中E(2) [LPF→σ*(F—Cl)]为17.204 kJ·mol-1,而CuCH2F…ClF、AgCH2F…ClF和AuCH2F…ClF的structure I复合物中E(2)[LPF→σ*(F—Cl)]分别为26.121、28.423和19.214 kJ·mol-1.从相对增加值来看,Ag系列复合物的E(2)增加的最多,Cu次之,Au列复合物的E(2)增加的最少.这样的结果与前面得到的键长、相互作用能和表面静电势的结果符合得非常好.为了更好地表达复合物之间的轨道相互作用,图4以CuCH2Br…ClF的两种复合物为例,给出了NBO图.从图中可以清晰地看到,在structure I复合物中,Br原子的孤对电子轨道与F—Cl键的反键轨道之间存在明显的轨道相互作用.而在structure II复合物中,除了这种相互作用外,Cl原子的孤对电子轨道与C—Cu键的反键轨道之间也有很强的作用,这与上文得到的二阶稳定化能是一致的.
从表3中可以看出,对于CuCH2X…ClF和AgCH2X…ClF的structure I复合物,当X原子变化时,相互作用能的数值变化不大.但是structure II复合物的情况明显不同:当X原子变化时,三种复合物相互作用能差距较大.例如CuCH2F…ClF(I)、CuCH2Cl…ClF(I)和CuCH2Br…ClF(I)的相互作用能分别是-18.870、-18.301和-18.896 kJ·mol-1,而CuCH2F…ClF(II)、CuCH2Cl…ClF(II)和CuCH2Br…ClF(II)的相互作用能则分别为-22.847、-27.062和-35.003 kJ·mol-1.这样的计算结果可以从二阶稳定化能方面得到很好的解释.如表4所示,上述三种Cu取代的structure I复合物的E(2)[LPX→σ*(F—Cl)]分别为26.121、37.507和49.939 kJ·mol-1,变化幅度不是很大.反观structure II复合物,对应的数值分别为31.018、69.236和149.817 kJ·mol-1,变化幅度非常大.另外,structure II复合物还有LPCl→σ*(C—M)的稳定作用.
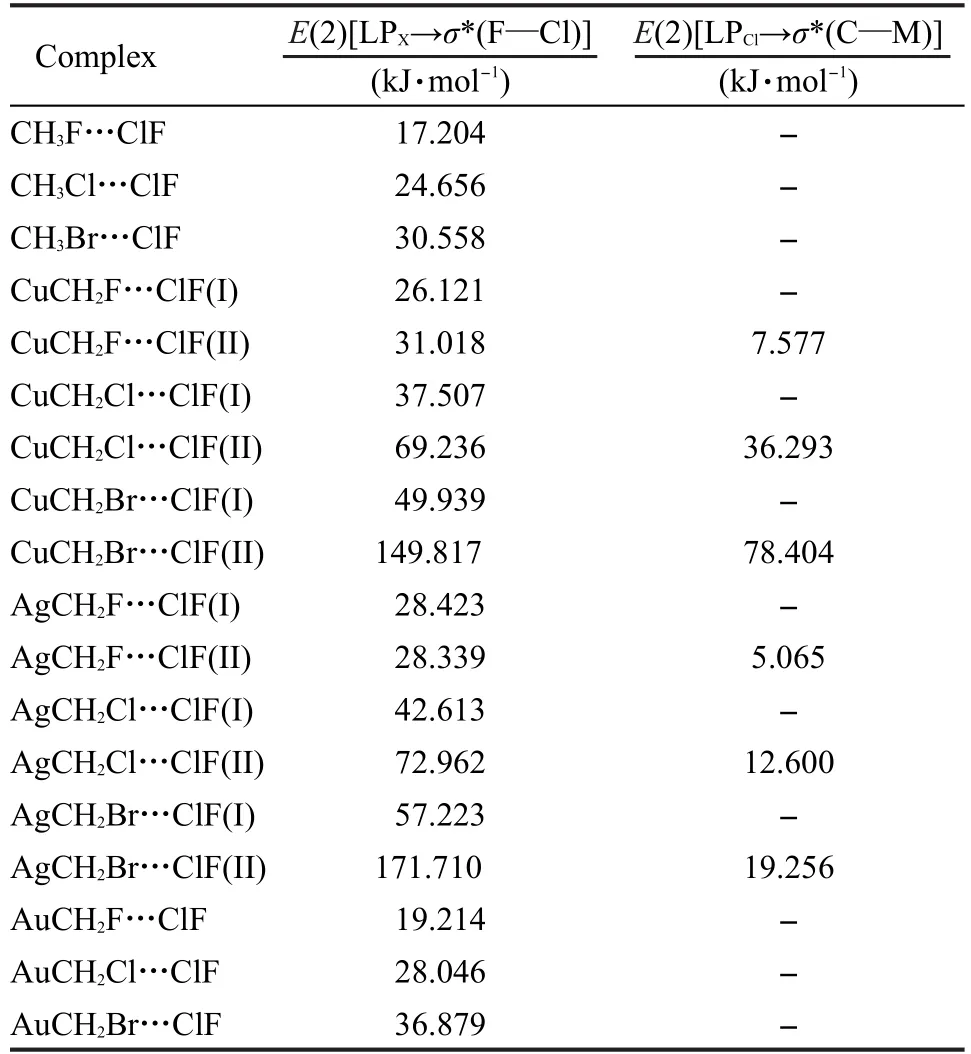
表4 CH3X···ClF和MCH2X···ClF复合物的二阶稳定化能Table 4 The second-order perturbation energies of the CH3X···ClF and MCH2X···ClF complexes

图4 CuCH2Br…ClF复合物的NBOs间的轨道相互作用Fig.4 Orbital interaction between the NBOs of the CuCH2Br…ClF complex(a)NBO interaction between LPBrand σ*(F—Cl)for the structure I complex of CuCH2Br…ClF,(b)NBO interaction between LPBr and σ*(F—Cl)for the structure II complex of CuCH2Br…ClF,(c)NBO interaction between LPCland σ*(C―Cu)
3.4 分子中的原子分析
AIM理论已经成为一种有效的工具来理解化学键的概念和化学键的强度.32由于现有的AIM软件都不能很好地支持赝势基组.因此我们只对CuCH2X…ClF复合物进行了分析.图5显示了CuCH2X…ClF复合物的AIM分子图,其中左边是structure I的分子图,右边是structure II的分子图.根据Bader的理论,32如果相邻的两个原子之间存在着成键作用,那么一定有一条从键鞍点(BCP)出发连接两原子的键径存在.从图5中可以看出,在structure I复合物中X原子与Cl原子之间有一个键鞍点且有两条键径分别与X原子和Cl原子相连.Structure II复合物的情况明显与structure I有所不同.在structure II复合物中,除了X原子与Cl原子之间的键鞍点外,Cu原子和Cl原子之间也有一个键鞍点.这个键鞍点的存在表明,除了卤键相互作用以外,还存在着另外一种弱相互作用,这与前面的讨论是吻合的.值得注意的是,structure II复合物中,还有一个由C、Cu、Cl和X原子组成的环鞍点,这个稳定点的出现也是复合物更加稳定的一个有力证据.
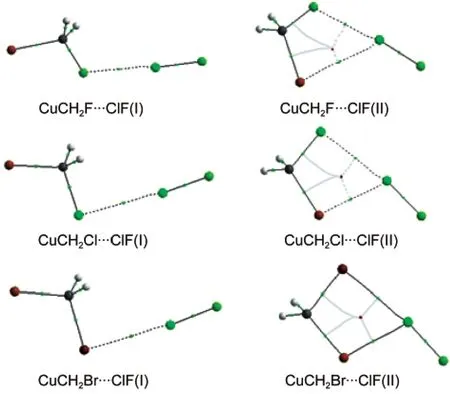
图5 CuCH2X…ClF复合物的分子图Fig.5 Molecular graphs for the CuCH2X…ClF complexes

表5 CuCH2X…ClF(structure I)复合物卤键键鞍点的拓扑性质(单位为a.u.)Table 5 Topological properties(in a.u.)for the critical point of the halogen bond in the CuCH2X…ClF (structure I)complex
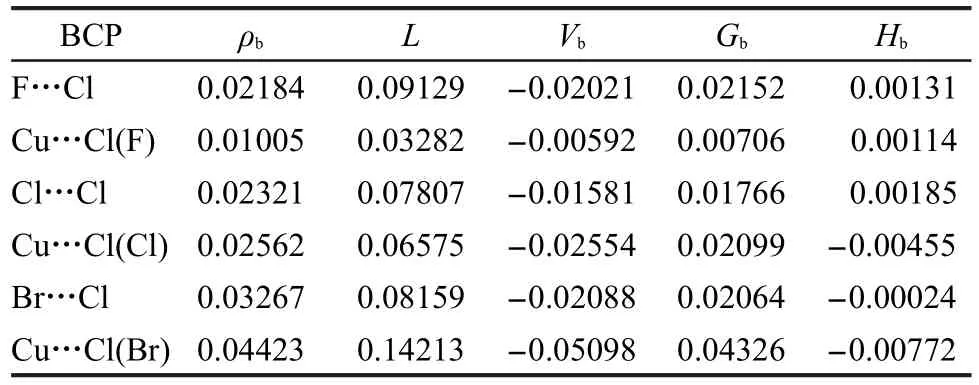
表6 CuCH2X…ClF(structure II)复合物键鞍点的拓扑性质(单位为a.u.)Table 6 Topological properties(in a.u.)for the CuCH2X…ClF(structure II)complex
为了更好地考察键鞍点的性质,表5和表6分别给出了structure I和II键鞍点的电子密度(ρb)、电子密度的拉普拉斯量(L)、动能密度(Gb)、势能密度(Vb)和能量密度(Hb).其中,Hb=Gb+Vb.Popelier33认为,氢键形成的条件是键鞍点的ρb在0.002-0.04 a.u.之间,L在0.024-0.139 a.u.之间.从表5和表6可以看出,不管是哪种类型的复合物,这两个条件都基本符合.能量密度Hb被认为是理解分子间相互作用的一个更准确的指标.当Hb大于零时,相互作用属于静电相互作用;当Hb小于零时,相互作用属于共价相互作用.Lu等34结合电子密度的拉普拉斯量,发展了一种新的卤键分类方法:弱卤键的L和Hb均为正;强卤键的L为正,Hb为负.从表6中可以看出,对于structure II来说,CuCH2F…ClF复合物的两个键鞍点的Hb皆为正;在CuCH2Cl…ClF复合物中,Cl…Cl之间的键鞍点的Hb为正,而Cu…Cl之间的键鞍点的Hb为负;CuCH2Br…ClF复合物的两个键鞍点的Hb皆为负.前文中的计算结果表明,structure II的CuCH2F…ClF、CuCH2Cl…ClF和CuCH2Br…ClF复合物的稳定性按照X原子的原子序数增大而逐渐加强,这与相应键鞍点处Hb的符号是相符的.
4 结论
采用量子化学方法系统研究了MCH2X…ClF (M=Cu,Ag,Au;X=F,Cl,Br)的卤键相互作用,并与不含有过渡金属的复合物CH3X…ClF进行了对比. CH3X…ClF复合物只有一种稳定结构,CH3X分子中的两个H原子朝向ClF分子的Cl原子.CuCH2X…ClF和AgCH2X…ClF复合物有两种稳定结构, AuCH2X…ClF复合物只有一种稳定结构.在发现的另一种稳定结构中,除了X…Cl之间的卤键相互作用外,还有M(M=Cu,Ag)…Cl之间的相互作用.
相对于CH3X…ClF复合物,MCH2X…ClF复合物中X…Cl之间的距离都有所缩短,稳定性也提高了.对于X原子相同的MCH2X…ClF复合物,Ag原子取代的增强效应最显著,Cu次之,Au最不明显.计算了MCH2X分子中X原子上的最负静电势,结果表明这些卤键相互作用与静电势的变化有一致的关系.运用NBO和AIM分析方法讨论了轨道相互作用、键鞍点以及键鞍点处的拓扑性质,得到的结果与相互作用距离和相互作用能一致.
(1) Jeffrey,G.A.An Introduction to Hydrogen Bonding;Oxford University Press:Oxford,1997.
(2) Sun,T.;Wang,Y.B.Acta Phys.-Chim.Sin.2011,27,2553. [孙 涛,王一波.物理化学学报,2011,27,2553.]
(3) Legon,A.C.Phys.Chem.Chem.Phys.2010,12,7746.
(4)Auffinger,P.;Hays,F.A.;Westhof,E.;Ho,P.S.Proc.Natl. Acad.Sci.U.S.A.2004,101,16789.
(5) Clark,T.;Hennemann,M.;Murray,J.S.;Politzer,P.J.Mol. Model.2007,13,291.
(6) Politzer,P.;Lane,P.;Concha,M.C.;Ma,Y.G.;Murray,J.S. J.Mol.Model.2007,13,305.
(7) Zhao,Y.;Zeng,Y.L.;Zhang,X.Y.;Zheng,S.J.;Meng,L.P. Acta Phys.-Chim.Sin.2006,22,1526.[赵 影,曾艳丽,张雪英,郑世钧,孟令鹏.物理化学学报,2006,22,1526.]
(8) Zhao,Y.;Zeng,Y.L.;Sun,Z.;Zheng,S.J.;Meng,L.P.Acta Phys.-Chim.Sin.2008,24,502.[赵 影,曾艳丽,孙 政,郑世钧,孟令鹏.物理化学学报,2008,24,502.]
(9)Yuan,K.;Liu,Y.Z.;Lü,L.L.;Ma,W.C.Acta Phys.-Chim.Sin. 2008,24,1257.[袁 焜,刘艳芝,吕玲玲,马伟超.物理化学学报,2008,24,1257.]
(10) Metrangolo,P.;Meyer,F.;Pilati,T.;Resnati,G.;Terraneo,G. Angew.Chem.Int.Edit.2008,47,6114.
(11) Metrangolo,P.;Neukirch,H.;Pilati,T.;Resnati,G.Accounts Chem.Res.2005,38,386.
(12) Metrangolo,P.;Resnati,G.Chem.Eur.J.2001,7,2511.
(13)Espallargas,G.M.;Brammer,L.;Allan,D.R.;Pulham,C.R.; Robertson,N.;Warren,J.E.J.Am.Chem.Soc.2008,130,9058.
(14) Smart,P.;Espallargas,G.M.;Brammer,L.Cryst.Eng. Commun.2008,10,1335.
(15) Brammer,L.;Espallargas,G.M.;Libri,S.Cryst.Eng.Commun. 2008,10,1712.
(16)Espallargas,G.M.;Zordan,F.;Marin,L.A.;Adams,H.; Shankland,K.;van de Streek,J.;Brammer,L.Chem.Eur.J. 2009,15,7554.
(17) Clemente-Juan,J.M.;Coronado,E.;Espallargas,G.M.;Adams, H.;Brammer,L.Cryst.Eng.Commun.2010,12,2339.
(18)Espallargas,G.M.;Brammer,L.;Sherwood,P.Angew.Chem. Int.Edit.2006,45,435.
(19) Zordan,F.;Espallargas,G.M.;Brammer,L.Cryst.Eng. Commun.2006,8,425.
(20) Brammer,L.;Espallargas,G.M.;Adams,H.Cryst.Eng. Commun.2003,5,343.
(21)Xu,L.;Lv,J.;Sang,P.;Zou,J.W.;Yu,Q.S.;Xu,M.B.Chem. Phys.2011,379,66.
(22) Bertani,R.;Sgarbossa,P.;Venzo,A.;Lelj,F.;Amati,M.; Resnati,G.;Pilati,T.;Metrangolo,P.;Terraneo,G.Coord. Chem.Rev.2010,254,677.
(23) Wachters,A.J.H.J.Chem.Phys.1970,52,1033.
(24)Hay,P.J.;Wadt,W.R.J.Chem.Phys.1985,82,270.
(25) Boys,S.F.Mol.Phys.1970,19,553.
(26) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision C.02;Gaussian Inc.:Wallingford,CT,2004.
(27) Reed,A.E.;Curtiss,L.A.;Weinhold,F.Chem.Rev.1988,88, 899.
(28) Keith,T.A.AIMAll,Version 10.05.04;TK Gristmill Software: Overland Park KS,USA,2010;http://aim.tkgristmill.com.
(29)Wang,F.F.;Hou,J.H.;Li,Z.R.;Wu,D.;Li,Y.;Lu,Z.Y. J.Chem.Phys.2007,126,144301.
(30) Tsipis,A.C.Organometallics 2010,29,354.
(31) Politzer,P.;Murray,J.S.;Clark,T.Phys.Chem.Chem.Phys. 2010,12,7748.
(32)Bader,R.F.W.Atoms in Molecules:a Quantum Theory;Oxford Unversity Press:Oxford,1990.
(33) Popelier,P.L.A.J.Phys.Chem.A 1998,102,1873.
(34)Lu,Y.X.;Zou,J.W.;Wang,Y.H.;Yu,Q.S.Int.J.Quantum Chem.2007,107,1479.
Influence of Transition Metal M(M=Cu,Ag,Au)on the Strength of Halogen Bonding Interaction X…Cl(X=F,Cl,Br)
ZHAO Qiang1,*FENG Da-Cheng2
(1Department of Chemical Engineering,Zibo Vocational Institute,Zibo 255314,Shandong Province,P.R.China;2Institute of Theoretical Chemistry,Shandong University,Jinan 250100,P.R.China)
Intermolecular complexes of MCH2X…ClF(M=Cu,Ag,Au;X=F,Cl,Br)and CH3X…ClF were investigated using by quantum chemistry method.Only one stable structure containing a halogen bond was obtained for the CH3X…ClF complexes.For the MCH2X…ClF complexes,as well as the halogen-bonded complex,another optimized structure containing both a halogen bond and M…Cl interaction was determined.The stability of the MCH2X…ClF complexes was greater than that of the CH3X…ClF complexes.Substitution with M improves the stability of the resulting complex with the order Ag>Cu>Au.The most negative molecular electrostatic potential of X in MCH2X and CH3X was calculated, and the decrease of this value is the main reason for the enhanced stability of these complexes.The characteristics of these complexes were also studied by natural bond orbital and atoms in molecules methods.The second-order perturbation energy and topological properties of the saddle points were calculated and the results were consistent with the interaction energy.
Halogen bond;Transition metal;Enhancing effect;Stability
10.3866/PKU.WHXB201203261
January 28,2012;Revised:March 23,2012;Published on Web:March 26,2012.
∗Corresponding author.Email:qzhaochem@yahoo.cn;Tel:+86-13864428619
O641
猜你喜欢
广州化工(2022年19期)2022-11-09 11:30:46
广州化工(2022年18期)2022-10-22 10:27:00
湘潭大学自然科学学报(2022年1期)2022-04-11 11:07:42
原子与分子物理学报(2021年2期)2021-03-29 07:30:46
硅酸盐通报(2020年1期)2020-02-25 10:01:30
中成药(2018年7期)2018-08-04 06:04:18
上海师范大学学报·自然科学版(2018年3期)2018-05-14 13:47:10
中成药(2018年3期)2018-05-07 13:34:18
经济数学(2017年4期)2018-01-18 17:25:55
纯粹数学与应用数学(2015年3期)2015-10-14 05:44:03
