
辛伐他汀对厄贝沙坦药代动力学影响的研究
2012-12-06 08:04周霞瑾齐惠珍牛哲哲王明霞
中国药理学通报 2012年5期
周霞瑾,齐惠珍,牛哲哲,王明霞,常 青
(1.河北医科大学第四医院药剂科,河北石家庄 050011;2.河北医科大学药学院,河北 石家庄 050017)
辛伐他汀是HMG-CoA(羟甲基戊二酸单酰辅酶A)还原酶抑制剂,在体内被水解成β-羟基酸代谢物,可降低血总胆固醇和低密度脂蛋白水平。也可中度降低血三酰甘油和升高高密度脂蛋白水平,是临床常用的调脂药[1]。高血脂能导致血管结构和功能异常,使血管舒张功能损伤[2],加重高血压病的危害,降血脂已成为高血压的防治原则之一。因此临床上他汀类降脂药常与厄贝沙坦等抗高血压药联用。有关二者的相互作用目前多限于药效学方面,而辛伐他汀对厄贝沙坦药代动力学的影响,目前尚未见报道,值得我们深入探讨。文献报道,辛伐他汀在肝脏主要经CYP3A4代谢[3],厄贝沙坦则主要经CYP2C9代谢,几乎不经CYP3A4代谢[4]。二者虽经不同酶代谢,但药物代谢酶的作用机制非常复杂。本课题研究了新西兰大白兔灌胃服用辛伐他汀不同时间后对厄贝沙坦的药代动力学的影响,旨在为临床药物相互作用提供理论参考。
1 仪器与试剂
1.1 仪器 Waters高效液相色谱仪,515泵,474荧光检测器,Empower色谱数据工作站;ZH-2涡旋混合器(天津药典标准仪器厂);800型离心机(上海分析仪器厂);梅特勒AE240电子分析天平(瑞士);SANYO低温冰箱。
1.2 药品和试剂 替米沙坦标准对照品(美国Sigma-Aidrich公司,Lot No T-8949);厄贝沙坦标准对照品(中国药品生物制品检定所,批号:100607-200301),乙腈为色谱纯,乙醚、二氯甲烷、磷酸为分析纯,试验用水为多效蒸馏水。羧甲基纤维素CMCS(天津市光复精细化工研究所批号:20050815,化学纯),辛伐他汀片(杭州默沙东制药有限公司,批号:100292,规格:20 mg/片),厄贝沙坦片[赛诺菲安万特(杭州)制药有限公司,批号:0A020,规格:0.15 g/片]。
1.3 动物 新西兰大白兔24只,均为♂,体质量2.5~3 kg,购于河北医科大学动物中心,动物合格证号为709198。
2 方法
2.1 动物处置 每只笼子装1只新西兰大白兔,称重,编号,进入动物室,经1周适应性饲养后,随机分为4组,分别为:3 d对照组、5 d对照组、辛伐他汀(5 mg·kg-1)诱导3 d组和辛伐他汀(5 mg·kg-1)诱导5 d组,每组6只。
3 d对照组:1~3 d每天1次灌胃给予2%羧甲基纤维素溶液,于d 4早8:00灌胃给予厄贝沙坦(50 mg·kg-1)的2%羧甲基纤维素溶液。
5 d对照组:1~5 d每天1次灌胃给予2%羧甲基纤维素溶液,于d 6早8:00灌胃给予厄贝沙坦(50 mg·kg-1)的2%羧甲基纤维素溶液。
辛伐他汀诱导3 d组:1~3 d灌胃给予辛伐他汀(5 mg·kg-1,qd)的2%羧甲基纤维素溶液,于d 4早8:00灌胃给厄贝沙坦(50 mg·kg-1)的2%羧甲基纤维素溶液。
辛伐他汀诱导5 d组:1~5 d灌胃给予辛伐他汀(5 mg·kg-1,qd)的2%羧甲基纤维素溶液,于d 6早8:00灌胃给厄贝沙坦(50 mg·kg-1)的2%羧甲基纤维素溶液。
所有动物于灌胃给予厄贝沙坦的前1 d晚8:00开始禁食不禁水,所有药物的给药剂量按人/兔体质量的等效剂量换算。
2.2 取血方法 预先为动物置留置针,分别于给药前和给药后 0.25、0.5、1、1.5、2、3、5、7、9、12、24、36 h从新西兰大白兔的耳中动脉取血2 ml,置肝素管中,3 000 r·min-1离心 10 min,取上清液,放入-80℃低温冰箱保存,用于测定厄贝沙坦的浓度。
2.3 血浆样品的处理 取血浆400 μl,置于10 ml具塞玻璃离心管中,精密加入内标工作液40 μl(5 mg·L-1),涡旋混匀 15 s,加入 100 μl磷酸溶液(1 mol·L-1),涡旋30 s,使其酸化,加入乙醚-二氯甲烷(7 ∶3)[5]混合液5 ml提取,涡旋振荡5 min,离心(1 500 r·min-1)5 min,取有机层4.5 ml于洁净干燥具塞玻璃离心管中,加入200 μl NaOH溶液(0.05 mol·L-1),涡旋震荡 3 min,离心(1 500 r·min-1)5 min,于-80℃低温冰箱中冷冻15 min,弃去上层未冻有机相,待下层融化后,用微量取样器取150 μl于1 ml的 Ephendroff管中,加入 100 μl磷酸溶液(0.2 mol·L-1)酸化,涡旋混匀 30 s,进样 50 μl分析。
2.4 色谱条件 色谱柱:Diamonsil C18柱(4.6 mm×200 mm,5 μm),流动相:水 - 乙腈(57 ∶43),三乙胺0.1%,磷酸调 pH为 3.00,流速:1.0 ml·min-1,柱温:室温;荧光检测波长:激发波长为254 nm,发射波长为 371 nm[6];内标物:替米沙坦;进样量:50 μl;该色谱条件下替米沙坦和厄贝沙坦的保留时间分别为4.2 min和6.0 min,分离效果良好,空白血浆的内源性物质对药物和内标物无干扰。
2.5 药代动力学分析
2.5.1 分析软件和方法 每只新西兰大白兔的血浆厄贝沙坦浓度值均用DAS version 3.0(Bontz Inc.,Beijing,China)药代动力学软件进行智能化数据处理,模拟血药浓度-时间曲线,并计算非房室模型统计矩参数。Cmax和Tmax为实测值。
2.5.2 数据处理和统计分析 统计分析采用SPSS13.0软件,计量资料采用±s,多组比较采用ANOVA,P<0.05时,再用 Dunnett检验;Tmax采用秩和检验。
3 结果
3.1 各组的平均血药浓度-时间曲线 见Fig 1和Fig 2。
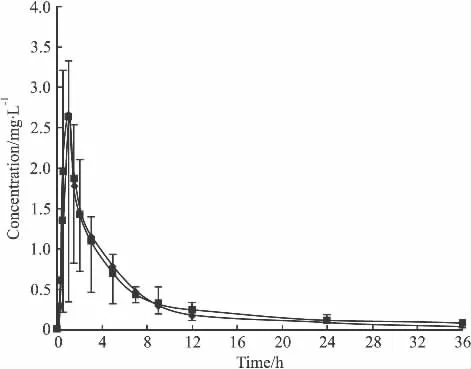
Fig 1 Mean plasma concentration-time curve of irbesartan in the control and simvastatin(5 mg·kg-1for 3 days)group(±s,n=6)
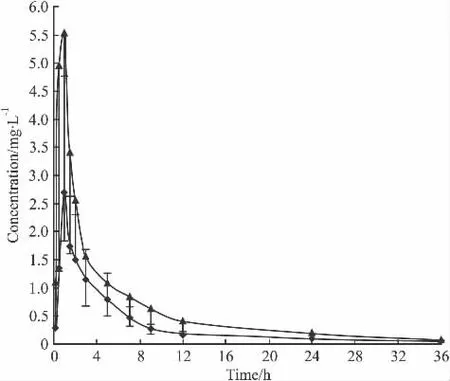
Fig 2 Mean plasma concentration-time curve of irbesartan in the control and simvastatin(5 mg·kg-1for 5 days)group(±s,n=6)
Tab 1 Mean noncompartment model's pharmacokinetic parameters of irbesartan after oral administration in the four groups(ig,50 mg·kg-1)(±s,n=6)
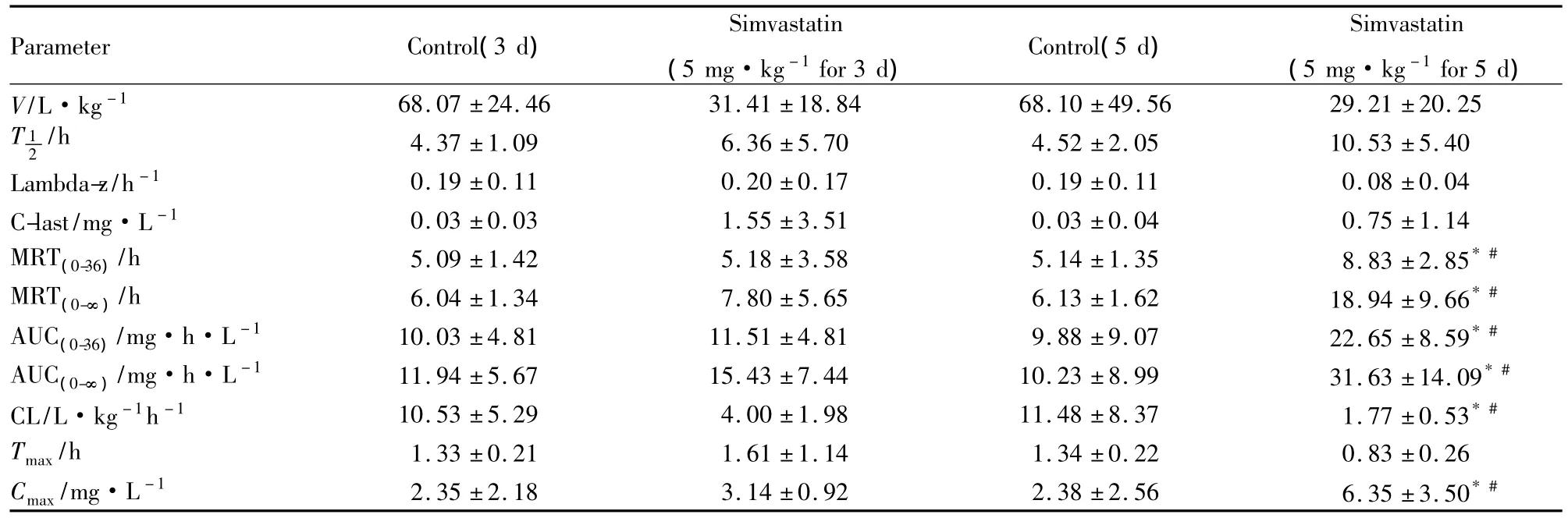
Tab 1 Mean noncompartment model's pharmacokinetic parameters of irbesartan after oral administration in the four groups(ig,50 mg·kg-1)(±s,n=6)
*P<0.05 vs control;#P<0.05 vs simvastatin for 3 d group
Parameter Control(3 d)Simvastatin(5 mg·kg-1for 3 d)Control(5 d)Simvastatin(5 mg·kg-1for 5 d)V/L·kg-1 68.07±24.46 31.41±18.84 68.10±49.56 29.21±20.25 T 12/h 4.37±1.09 6.36±5.70 4.52±2.05 10.53±5.40 Lambda-z/h-1 0.19±0.11 0.20±0.17 0.19±0.11 0.08±0.04 C-last/mg·L-1 0.03±0.03 1.55±3.51 0.03±0.04 0.75±1.14 MRT(0-36)/h 5.09±1.42 5.18±3.58 5.14±1.35 8.83±2.85*#MRT(0-∞)/h 6.04±1.34 7.80±5.65 6.13±1.62 18.94±9.66*#AUC(0-36)/mg·h·L-1 10.03±4.81 11.51±4.81 9.88±9.07 22.65±8.59*#AUC(0-∞)/mg·h·L-1 11.94±5.67 15.43±7.44 10.23±8.99 31.63±14.09*#CL/L·kg-1h-1 10.53±5.29 4.00±1.98 11.48±8.37 1.77±0.53*#Tmax/h 1.33±0.21 1.61±1.14 1.34±0.22 0.83±0.26 Cmax/mg·L-1 2.35±2.18 3.14±0.92 2.38±2.56 6.35±3.50*#
3.2 药代参数 各组的血药浓度-时间数据经DAS 3.0软件处理后的非房室模型统计矩参数见Tab 1。
经单因素方差分析,辛伐他汀诱导3 d组与对照组比较,厄贝沙坦的主要药动学参数差异无统计学意义;辛伐他汀诱导5 d组的药代动力学参数,与对照组比较有统计学差异(P<0.05),AUC(0-36)、AUC(0-∞)、MRT(0-36)、MRT(0-∞)和Cmax均升高 (P<0.05),而CL降低(P<0.05);辛伐他汀诱导5 d组与3 d组相比,厄贝沙坦的主要药代动力学参数差异有 显 著 性 (P< 0.05),AUC(0-36)、AUC(0-∞)、MRT(0-36)、MRT(0-∞)和Cmax均升高(P<0.05),而 CL降低(P<0.05),各组Tmax变化不大,经秩和检验差异无统计学意义。
4 讨论
文献报道,家兔取血多采用耳缘静脉取血和心脏取血。我们实验中发现,由于药代动力学研究中口服给药吸收相的取血点相对密集,耳缘静脉血难以满足要求的取血量。而心脏取血因不能连续取血,不适用于药动学研究。本实验中,我们采用在兔耳中动脉埋留置针取血,方法方便、快捷,血量充足,能满足大多数药代动力学的研究。
在本实验中,经辛伐他汀(5 mg·kg-1)诱导5 d 后,厄贝沙坦的 AUC(0-36)、AUC(0-∞)和Cmax均升高,清除率CL则由11.487±8.367降为1.773±0.531,减少了约5倍,具有统计学意义(P<0.05),表明辛伐他汀对厄贝沙坦的清除率亦具有明显影响。而且厄贝沙坦的体内滞留时间MRT亦延长(P<0.05),与AUC升高、CL减少一致。辛伐他汀是CYP3A4的特异性底物[3],而厄贝沙坦主要经CYP2C9氧化,几乎不被 CYP3A4代谢[4]。因此我们认为辛伐他汀对厄贝沙坦药代动力学的明显影响应与CYP2C9无关,而可能涉及CYP3A酶和P-糖蛋白。
众所周知 P-糖蛋白(P-glycoprotein,P-gp)是人体药物处置中最重要的药物转运体,它可利用ATP水解释放的能量将作用底物从细胞内转运至细胞外。在人体正常组织肝脏、肾脏、肠道、胎盘、血脑屏障、血睾屏障以及淋巴细胞系和心脏内小动脉、毛细血管等部位都有分布。P-gp在人体正常组织内的分布以及对药物的逆向转运功能使得P-gp在药物的吸收、分布、代谢和清除方面具有重要意义[7]。
P-gp的作用底物范围非常广泛,底物的亲脂性可能是决定它与P-gp结合的最重要参数。厄贝沙坦的脂溶性决定其可能为P-gp底物。Bourrié等[8]在研究中首先发现厄贝沙坦是P-gp的底物。Weiss等[9]在研究血管紧张素受体Ⅰ拮抗剂与ATP结合转运体的相互作用时,也发现厄贝沙坦对P-gp有弱抑制作用。
已知P-gp和CYP3A4的底物有重叠,辛伐他汀是CYP3A4的底物。Bogman等[10]证实他汀类如辛伐他汀、阿托伐他汀和洛伐他汀在高浓度时抑制P-gp。Wang等[11]也证实辛伐他汀、洛伐他汀和阿托伐他汀直接抑制P-gp。Sieczkowski等[12]发现辛伐他汀可能通过损坏P-gp的糖基化而抑制P-gp的功能,并且损坏程度随使用时间延长而增加,与本实验结果一致。即与辛伐他汀诱导3 d组比,辛伐他汀诱导5 d组影响厄贝沙坦的药代动力学。
综上所述,辛伐他汀对厄贝沙坦药代动力学的影响可能与辛伐他汀和厄贝沙坦同为药物转运体P-gp的抑制剂有关,二者竞争性抑制P-gp,从而引起厄贝沙坦药动学参数的改变。有关辛伐他汀对厄贝沙坦的药代动力学影响的P-gp作用机制有待今后做进一步研究。
总之,本研究结果显示,辛伐他汀诱导5 d后对兔体内厄贝沙坦的主要药代动力学参数有明显影响,其影响机制可能与P-gp有关。
[1]Kashani A,Phillips C O,Foody J M,et al.Risks associated with statin therapy:a systematic overview of randomized clinical trials[J].Circulation,2006,114:2788-97.
[2]孔 祥,杨解人,郭莉群,等.单纯肾性和复合型肾性高血压大鼠主动脉功能的对比研究[J].中国药理学通报,2009,25(2):252-5.
[2]Kong X,Yang J R,Guo L Q,et al.Comparing study of aortic function between renal hypertension rat and renal hypertensive-hyperlipidemia rat?[J].Chin Pharmacol Bull,2009,25(2):252-5.
[3]Foti R S,Rock D A,Wienkers L C,et al.Selection of alternative CYP3A4 probe substrates for clinical drug interaction studies usingin vitrodata andin vivosimulation[J].Drug Metab Dispos,2010,38:981-7.
[4]Taavitsainen P,Kiukaanniemi K,PelkonenO.In vitroinhibitionscreening of human hepatic P450 enzymes by five angioten-sin-Ⅱreceptor antagonists[J].Clin Pharmacol,2000,56:135-40.
[5]Shakya A K,Al-Hiari Y M,Alhamami O M.Liquid chromatographic determination of irbesartan in human plasma[J].Chromatogr B Analyt Technol Biomed Life Sci,2007,848(2):245.
[6]Gonzalez L,Lopez J A,Alonso R M,et al.Fast screening method for the determination of angiotensin receptor antagonists in human plasma by high-performance liquid chromatography with fluorimetric detection[J].Chromatogr A,2002,949(1-2):49-60.
[7]Erb R.Implications of genetic polymorphisms in drug transporters for pharmacotherapy[J].Cancer Lett,2006,234(1):4-33.
[8]Bourrié M,Meunier V,Berger Y,et al.Role of cytochrome P4502C9 in Irbesartan oxidation by human liver microsomes[J].Drug Metab Dispos,1999,27(2):288-96.
[9]Weiss J,Sauer A,Divac N,et al.Interaction of angiotensin receptor typeⅠ blockers with ATP-binding cassette transporters[J].Biopharm Drug Dispos,2010,31(2-3):150-61.
[10]Bogman K,Peyer AK,Török M,et al.HMG-CoA reductase inhibitors and P-glycoprotein modulation[J].Pharmacol,2001,132:1183-92.
[11]Wang E,Casciano C N,Clement R P,Hohenegger M.HMG-CoA reductase inhibitors(statins)characterized as direct inhibitors of P-glycoprotein[J].Pharm Res,2001,18:800-6.
[12]Sieczkowski E,Lehner C,Ambros P F,Hohenegger M.Double impact on p-glycoprotein by statins enhances doxorubicin cytotoxicity in human neuroblastoma cells[J].Int J Cancer,2010,126(9):2025-35.
猜你喜欢
化工管理(2022年14期)2022-12-02
昆明医科大学学报(2021年6期)2021-07-31
中华养生保健(2020年10期)2021-01-18
造纸化学品(2019年6期)2020-01-17
化工管理(2020年5期)2020-01-15
天然产物研究与开发(2019年1期)2019-03-01
中国造纸学报(2017年4期)2018-01-03
中国医药指南(2017年3期)2017-11-13
转化医学电子杂志(2015年4期)2015-12-27
中国药业(2014年21期)2014-05-26
