
转化生长因子β1与癫痫
2012-12-06 08:03李良勇
中国药理学通报 2012年5期
李良勇,王 玉
(1.安徽中医学院第一附属医院脑病中心,安徽合肥 230031;2.安徽医科大学第一附属医院神经内科,安徽合肥 230032)
癫痫是神经系统的常见病之一,具有反复发作及不可预测的特点,癫痫的发病率为人群的1‰,年患病率为5‰~7‰,我国估计难治性癫痫患者不少于100万[1],因此进一步探索癫痫发病机制,寻找新的防止癫痫发生及控制癫痫发作的手段十分必要。转化生长因子 TGF-β1(transforming growthfactorbeta1)是一种多功能细胞因子,具有广泛性生物学作用,系列研究证实,TGF-β1与神经系统的许多生理、病理过程有关[2-3],并发挥着神经保护作用。随着研究的深入,人们发现TGF-β1与癫痫有着密切的关系,已成为神经科学界研究的热点,本文就TGF-β1在癫痫发病中的作用、涉及的可能的机制及相关的信号转导途径进行综述。
1 TGF-β的生物学特性及信号传导
TGF-β是一种多功能细胞因子,由多种组织细胞合成,以自分泌、旁分泌和内分泌的方式,通过细胞表面的受体信号转导途径调控着细胞的增殖、分化、黏附、转移和凋亡,并参与细胞外基质(extracellular matrix,ECM)的合成以及血管生成等生理学过程,在涉及纤维化、损伤修复、骨骼重建和肿瘤生长等的许多病理学过程中发挥重要作用。活性TGF-β是由二硫键连接的两个各含112个氨基酸亚单位组成的二聚体多肽,分子量为25 kDa。TGF-β共有5种异构体,哺乳动物的 TGF-β 有 TGF-β1,TGF-β2,TGF-β3 三种亚单位,具有高度的同源性,三者在体外的作用相似,但在体内的生物学特性却不尽相同[2-4]。
TGF-β1和相关因子的信号转导是通过细胞表面的TGF-β受体介导的。TGF-β的信号转导始于TGF-β与细胞表面Ⅱ型受体上的二聚体结合形成复合体,然后I型受体识别并结合该复合体,形成I型受体一配体一Ⅱ型受体三聚体复合体。在此过程中,Ⅱ型受体胞内结构域的Ser/T-hr蛋白激酶结构域通过使I型受体胞内GS结构域磷酸化而激活I型受体,活化的I型受体再磷酸化,进一步作用于胞内下游分子Smads蛋白,激活的Smads蛋白进入细胞核,和其它的核协同或抑制因子结合,调节目标基因转录,使TGF-β信号转导人胞质。Smad家族是最早被证实的TGF-β受体激酶的底物,存在于细胞质中,可将信号由胞膜直接传至胞核。细胞内至少存在Smad(l-9)9种分子[2-3]。
细胞内至少存在Smadl-9 9种分子,按结构和功能可分为3个亚型:① 受体激活型Smad(receptor-activatedSmad,RSmad),包括 Smad 1、2、3、5、8、9,其中 Smad2、3 则可被 TGF-β激活,作为TGF-βRI激酶的直接底物,活化后与通用型Smad(Co-Smad)形成复合物异位人核,转导特异性信号,调节靶基因的转录;② 通用型Smad(common-partnerSmad,Co-Smad),哺乳动物中仅有Smad4,能与活化的R-Smad形成异聚体,在信号转导中起瓶颈作用;③ 抑制型Smad(inhibitory Smad,I-Smad),包括 Smad6、7,可阻断受体介导的 R-Smad磷酸化,阻碍活化的R-Smad与Co-Smad异聚体的形成,还可与活化的受体或受体激活的Smads竞争性结合形成无活性复合物,从而对R-Smad和Co-Smad介导的基因表达产生抑制作用[2-3]。
2 TGFβ1与癫痫
2.1 TGF-β1在癫痫中的正面作用及其相关机制分析Morgan[5]等用海仁藻酸(KA)诱导海马培养细胞边缘发作(limbic seizure),研究发现注射KA 4 h后受损的神经元中TGFβ1mRNA表达增高,2 d后达到高峰。体内TGFβ1缺乏的转基因鼠(TGFβ1-/+),注射KA后,癫痫发作更明显,神经元损伤更严重,并出现广泛的神经元凋亡现象[6]。杏仁核点燃大鼠模型中TGFβ1mRNA表达水平比对照组明显增高[7]。由此,在癫痫发作时TGFβ1可能具有潜在的神经保护作用。且近年来有研究报道,脑缺血缺氧参与了匹罗卡品诱导的SE后选择性神经元损伤机制[8]。而TGFβ1在缺血缺氧脑损伤中具有神经保护作用[9],由此,我们推测TGFβ1对SE后海马神经元损伤神经保护作用可能与前者有着相似的机理。其机制可能涉及以下几个方面:①既往研究表明TGFβ1能够降低海马神经元钙离子浓度,防止神经元内钙离子超载,维持神经元钙离子稳态[10]。而钙稳态失调,可使细胞内钙流增加引起神经元兴奋,且钙稳态失调亦可通过影响神经递质释放引起兴奋抑制功能失调并可以触发一系列反应并最终导致神经元死亡从而参与癫痫发生[11]。② Prehn把培养的鼠脑细胞置于含1 mML谷氨酸的无血清培养基中,在加入谷氨酸后立刻给予TGFβ1,18 h后发现,1~10 ng的TGFβ1可明显减轻谷氨酸的毒性;且进一步研究发现,TGFβ1能拮抗谷氨酸盐的神经毒性作用,下调NMDA受体的过度活化[12]从而发挥神经保护作用。谷氨酸是脑内重要的兴奋性递质,其能够激活NMDA受体,产生神经毒性并可触发癫痫发作[11]。③ TGFβ1通过上调 bcl-2(抗凋亡蛋白),抑制caspase-3(促凋亡蛋白)等内源外源性途径抑制癫痫神经元的凋亡[13]。癫痫持续状态(status epilepticus,SE)可导致海马等结构的神经元选择性死亡或凋亡,且这种选择性神经元死亡又能促进癫痫的形成和进展[11]。我们的研究已经证实外源性TGFβ1在癫痫大鼠中具有抗凋亡等神经元保护作用(文章待发表)。
TGFβ1还可通过上调靶细胞星形胶质细胞PAI-l表达和抑制tPA引起的神经元钙离子内流增加来保护神经元[14]。tPA(组织型纤溶酶原激活剂)可通过NMDA受体诱导神经元钙离子内流,其机制与tPA裂解NMDA受体的NRI亚基有关,而钙离子内流增加可以触发一系列反应并最终导致神经元死亡。Pal-1(纤溶酶原激活剂抑制剂-1)是tPA的天然抑制剂,tPA/PAI-1轴在NAMD介导的兴奋性毒性中起重要作用[14]。不仅如此,TGFβ1可通过抑制靶细胞小胶质细胞中的氧自由基而发挥作用[15]。上述的机制推测将是以后我们实验进一步研究的方向。
2.2 TGF-β1在癫痫中的负面作用及其相关机制分析
2.2.1 TGFβ1在癫痫中的负面作用 Ivans等破坏鼠血脑屏障建立脑损伤模型,或把白蛋白直接暴露于大脑新皮层,导致皮层功能紊乱,并记录到皮层癫痫样放电。深入研究发现白蛋白由TGF-βR(受体)介导被星形胶质细胞吸收,引起星形胶质细胞钾离子通道内部整流功能下降和细胞外高钾,导致NMDA受体过度活化,甚至癫痫样放电。且指出白蛋白被星形胶质细胞而非神经元吸收,且运用TGF-βR阻断剂可显著减少癫痫样放电[16]。后Ivans又进一步研究发现破坏血脑屏障暴露TGF-β1,产生的现象和上述一样。即TGF-βR介导TGF-β1被星形胶质细胞吸收产生癫痫样放电。因TGFβ1信号转导时TGFβ1首先与细胞膜表面的TGFβRⅡ二聚体结合,形成二元复合物,自动磷酸化时激活TGFβRⅠ,活化的TGFβRⅠ在进一步作用于细胞内的下游分子Smads蛋白,使TGFβ1的信号向细胞内转导。于是研究者认为白蛋白由TGFβRⅡ介导被星形胶质细胞吸收,并提示TGFβ1号转导途径可能参与了癫痫的形成机制[17]或TGFβ1可通过相关信号转导途径形成癫痫样放电。癫痫具有反复发作的特点,而我们的研究发现外源性TGFβ1可明显抑制SE大鼠的自发性发作的次数和程度,并可发挥抗海马神经元凋亡等神经保护作用(文章待发表),从而提示TGFβ1可通过另一信号转导途径抑制癫痫的自发反复性发作。“癫痫耐受”最早由Kelly&Mclntyre提出,相关机制的研究已成为研究热点并逐渐在各种动物模型中得到揭示。即一次或短暂的痫性发作诱导脑组织启动内源性保护机制,提高脑组织对后续较严重的SE的耐受能力,可以表现为神经元凋亡或坏死减少,癫痫发作次数减少程度减轻,甚至可能影响癫痫的形成和发展[18-19]。上述TGFβ1在星形胶质细胞中介导的信号传导路径可能在癫痫模型中所产生的复杂的神经网络机制中诱发内源性的保护机制,如上调NF-κB、Bcl-2或激发其它保护性的信号转导路径等,从而影响癫痫的形成和发展。
2.2.2 TGFβ1在癫痫中的信号转导途径 通过大量的文献及研究对上述的可能的信号转导路径总结分析如下。①TGFβ1在信号转导过程中激活两种不同的 TGFβRⅠ即ALK1(activin-like kinase receptor 1)和ALK5;ALK1和ALK5又分别进一步激活下游分子Smad1/5和Smad2/3即ALK1/Smad1/5和ALK5/Smad2/3转导途径。深入研究发现,在脑损伤时,TGFβ1可通过ALK1/Smad1/5介导激活神经元核因子NF-κB,核因子NF-κB具有抗凋亡等神经保护作用。虽在海马神经元受损时ALK5/Smad2/3同时被激活,但相对于ALK1/Smad1/5,前者表达水平低,持续时间短暂。在神经元中,核因子 NF-κB 可被 TGFβ1 通过 ALK1(TGF-βRⅠ)/Smad1/5介导激活,神经元核因子NF-κB又可上调Smad7的表达水平。我们知道生理状态下Smad7阻碍Smad2/3的磷酸化,抑制TGFβ的信号传递。因此我们可以推测在神经元中Smad7可能抑制ALK5从而抑制Smad2/3的磷酸化,正由于Smad7的抑制,致使ALK5在神经元中低表达,ALK1高表达,其信号传递占优势地位。由此认为,TGFβ1/ALK1/Smad1/5信号转导路径主要存在于神经元中并发挥神经保护作用[20-21](如Fig 1)。② 同时研究发现,ALK5/Smad2/3主要存在于星形胶质细胞中,而ALK1/Smad1/5在星形胶质细胞中相对于前者表达水平低,持续时间短暂[20-21]。联系上述实验,TGF-βR介导TGF-β1被星形胶质细胞吸收产生癫痫样放电,我们可以提出假设其机制可能是ALK5(TGF-βRⅠ)/Smad2/3介导TGF-β1或白蛋白在星形胶质细胞中产生作用被胶质细胞吸收而诱发癫痫样放电。Wang等从30名颞叶癫痫患者脑组织检测到TGF-βRⅠ的蛋白表达比对照组显著增高,且利用免疫荧光技术发现TGF-βRⅠ主要聚集在星形胶质细胞细胞质中[22]。提示 TGF-β1激活的ALK5(TGF-βRⅠ)/Smad2/3信号转导路径在星形胶质细胞中的优势地位及对癫痫的可能的促进作用。(如Fig 1)。在内皮细胞中,ALK5促进ALK1的表达,ALK1抑制 ALK5的信号传递,因而ALK1和ALK5两者的传导路径之间相平衡[23]。由于TGFβ1生物学性质受细胞类型影响,由此我们可以推测在星形胶质细胞中可能与内皮细胞相反,ALK1促进ALK5的表达,ALK5抑制ALK1的信号传递,从而导致星形胶质细胞中ALK5高表达,其信号传递占优势地位。
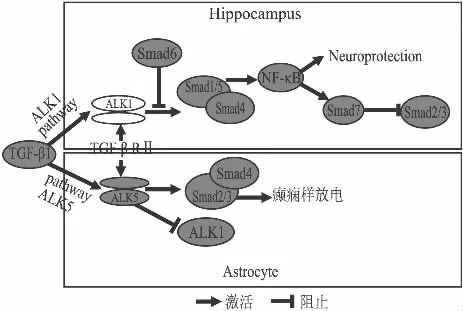
Fig 1 Signal transduction pathway of TGFβ1 in hippocampus and astrocyte
因此,TGFβ1信号传导路径对癫痫可能有着负面作用,尤其TGFβ1受体ALK5(TGF-βRⅠ)在星形胶质细胞中的作用可能会引发癫痫样放电。而将谷氨酸受体激动剂NMDA作用于海马培养细胞,König等发现TGFβ1通过ALK1(TGF-βRⅠ)激活神经元核因子NF-κB发挥抗凋亡等神经保护作用[20];TGFβ1还可通过上调星形胶质细胞PAI-l表达和抑制tPA引起的神经元钙离子内流增加来保护神经元,并抑制神经细胞凋亡[14]。由此,TGFβ1对神经元细胞和星型胶质细胞产生的作用不同。既往研究已表明,TGFβ1的调节作用因细胞类型、刺激物和实验条件不同而不同[2,17,24]。Ariane de Luca等报道TGFβ1诱导低剂量钾离子培养的非成熟小脑颗粒细胞凋亡,在高剂量钾离子培养的非成熟小脑颗粒细胞中则无作用[24]。但在复杂的癫痫神经网络中,各种信号转导路径同处于一个有机整体,“癫痫耐受”的机制可能参与其中。
近些年来越来越多的研究发现TGF-β1在中枢神经系统疾病中的神经保护作用,可能会为TGF-β1和癫痫的研究提供一些新的思路。以期为癫痫的防治提供新策略,为寻找癫痫的药物治疗提供新靶点。
[1]臧颖卓,范亚林,李 虹.癫痫发病机制的研究现状[J].脑与神经疾病杂志,2009,17(1):78-80
[1]Zang Y Z,Fan Y L,Li H.Study of epileptic pathogenesis[J].J Brain Nerv Dis,2009,17(1):78-80.
[2]Santibañez J F,Quintanilla M,Bernabeu C.TGF-β /TGF-β receptor system and its role in physiological and pathological conditions[J].Clin Sci,2011,121(6):233-51.
[3]Moustakas A,Heldin C H.The regulation of TGFβ signal transduction[J].Development,2009,136(22):3699-714.
[4]刘 浩,巍 伟.TGFβ 信号转导通路及以其为靶点的肝纤维化治疗[J].中国药理学通报,2007,23(5):561-5.
[4]Liu H,Wei W.TGFβ signal pathway and anti TGFβ strategies for treatment of liver fibrosis[J].Chin Pharmacol Bull,2007,23(5):561-5.
[5]Morgan T E,Nichols N R,Pasinetti G M,et al.TGF-β1mRNA increase in macrophage/microglial cell of the hippocampus in response to deafferentation and kainic acid-induced neurodegeneration[J].Exp Neurol,1993,120(2):291-301.
[6]Brionne T C,Tesseur I,Masliah E,et al.Loss of TGF-β1 Leads to Increased Neuronal Cell Death and Microgliosis in Mouse Brain[J].Neuron,2003,40(6):1133-45.
[7]Plata-Salamán C R,Ilyin S E,Turrin N P,et al.Kindling modulates the IL-1β system,TNF-α,TGF-β1,and neuropeptide mRNAs in specific brain regions[J].Brain Res Mol Brain Res,2000,75(2):248-58.
[8]Fabene P F,Merigo F,Galiè M,et al.Pilocarpine-induced status epilepticus in rats involves ischemic and excitotoxic mechanisms[J].PLoS One,2007,2(10):e1105.
[9]Ma M,Ma Y,Yi X,et al.Intranasal delivery of transforming growth factor-beta1 in mice after stroke reduces infarct volume and increases neurogenesis in the subventricular zone[J].BMC Neurosci,2008,9(117):1-10.
[10]Prehn J H,Bindokas V P,Marcuccilli C J,et al.Regulation of neuronal Bcl2 protein expression and calcium homeostasis by transforming growth factor type β confers wide-ranging protection on rat hippocampal neurons[J].Proc Natl Acad Sci USA,1994,91(26):12599-603.
[11]Scorza F A,Arida R M,Naffah-Mazzacoratti Mda G,et al.The pilocarpine model of epilepsy:what have we learned[J]?.An Acad Bras Cienc,2009,81(3):345-65.
[12]P rehn J H,Backhauss C,Krieg J,et al.Transforming growth factor betal prevents glutamate neurotoxicity in rat neocrtical cultures and protects mouse neocortes from ischemic injuryin vivo[J].J Cereb Blood Flow Metab,1993,13(3):521-5.
[13]Zhu Y,Ahlemeyer B,Bauerbach E,et al.TGF-β1 inhibits caspase-3 activation and neuronal apoptosis in rat hippocampal cultures[J].Neurochem Int,2001,38(3):227-35.
[14]Buisson A,Lesne S,Docagne F,et al.Transforming Growth Factor-β and Ischemic Brain Injury[J].Cell Mol Neurobiol,2003,23(4-5):539-50.
[15]Qian L,Wei S J,Zhang D,et al.Potent anti-inflammatory and neuroprotective effects of TGF-beta1 are mediated through the inhibition of ERK and p47phox-Ser345 phosphorylation and translocation in microglia[J].J Immunol,2008,181(1):660-8.
[16]Ivens S,Kaufer D,Flores L P,et al.TGF-β receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis[J].Brain,2007,130(2):535-47.
[17]Cacheaux L P,Ivens S,David Y,et al.Transcriptome Profiling Reveals TGF-β Signaling Involvement in Epileptogenesis[J].J Neurosci,2009,29(28):8927-35.
[18]Jimenez-Mateos E M,Hatazaki S,Johnson M B,et al.Hippocampal transcriptome after status epilepticus in mice rendered seizure damage-tolerant by epileptic preconditioning features suppressed calcium and neuronal excitability pathways[J].Neurobiol Dis,2008,32(3):442-53.
[19]Jimenez-Mateos E M,Henshall D C.Seizure preconditioning and epileptic tolerance:models and mechanisms[J].Int J Physiol Pathophysiol Pharmacol,2008,1(2):180-91.
[20]König H G,Kögel D,Rami A,et al.TGF-β1 activates two distinct type I receptors in neurons:implications for neuronal NF-κB signaling[J].J Cell Biol,2005,168(7):1077-86.
[21]Friedman A,Kaufer D,Heinemann U.Blood-brain barrier breakdown-inducing astrocytic transformation:Novel targets for the prevention of epilepsy[J].Epilepsy Res,2009,85(2-3):142-9.
[22]Lu Y,Xue T,Yuan J,et al.Increased expression of TGFβ type I receptor in brain tissues of patients with temporal lobe epilepsy[J].Clin Sci,2009,117(1):17-22.
[23]Goumans M J,Valdimarsdottir G,Itoh S,et al.Activin receptorlike kinase(ALK)1 is antagonistic mediator of lateral TGFβ/ALK5 signaling[J].Mol Cell,2003,12(4):817-28.
[24]Sanchez-Capelo A.Dual role for TGF-beta1 in apoptosis[J].Cytokine Growth Factor Rev,2005,16(1):15-34.
猜你喜欢
传染病信息(2022年2期)2022-07-15
湖南农业大学学报(自然科学版)(2021年4期)2021-08-14
昆明医科大学学报(2021年3期)2021-07-22
云南医药(2021年3期)2021-07-21
神经损伤与功能重建(2020年10期)2020-12-23
神经损伤与功能重建(2020年11期)2020-12-01
初中生世界·七年级(2018年7期)2018-09-07
江苏农业科学(2017年14期)2017-10-10
中国病理生理杂志(2015年8期)2015-12-21
中国药理学与毒理学杂志(2015年3期)2015-12-16
