
含羧酸镁(钙)官能团的多孔芳香骨架材料储氢性能的预测
2012-11-30 10:33:28苗延霖孙迎新
物理化学学报 2012年3期
苗延霖 孙 淮 王 琳 孙迎新
(上海交通大学化学化工学院,上海200240)
含羧酸镁(钙)官能团的多孔芳香骨架材料储氢性能的预测
苗延霖 孙 淮*王 琳 孙迎新
(上海交通大学化学化工学院,上海200240)
用MP2方法,TZVPP基组以及基组重叠误差(BSSE)校正计算了氢分子与修饰在多孔芳香骨架(PAF)上的羧酸镁、羧酸钙官能团的相互作用,并建立了描述这一相互作用的分子力学力场.在此基础上用巨正则系综蒙特卡洛(GCMC)模拟预测了氢气在该种新型PAF材料上的吸附等温线.量子化学计算结果表明,每个羧酸镁、羧酸钙官能团分别可以提供13、14个氢分子吸附位点,与每个氢分子的平均结合能在8 kJ·mol-1左右.通过比较不同温度和压力下材料的绝对吸附量和超额吸附量发现,在PAF骨架中引入羧酸镁、羧酸钙官能团可以显著提高材料的综合储氢性能,达到并超过了美国能源部提出的2015年储氢标准.同时该工作还揭示了氢吸附量与材料的表面积、空腔体积和分子作用强度间的复杂关系.
储氢;二羧酸盐;多孔芳香骨架;从头算;分子模拟
1 引言
氢能开发和利用的研究任务之一是开发高效、安全的储氢材料.在物理储氢方面,金属有机骨架(MOF)材料因合成容易,可以通过组合不同的连接基团构造,具有很高自由体积以及表面积的特点而受到广泛关注.1-7在低温77 K,5.6 MPa条件下,现有的MOF材料储氢量可以达到9.05%(质量分数,下同).8但目前合成得到的MOF材料在近室温条件下还仍未达到可以实际应用的目标.
近年来,多孔芳香骨架(PAF)材料的提出为近室温下达到高储氢量的目标提供了基础.9从预测结果来看,PAF-3(3表示骨架的连接体由3个苯环构成)的氢气吸附量很大,是一类极具潜力的储氢材料.但是,PAF材料的共价键本质及其过大的自由体积决定了它与氢分子的相互作用较弱,在室温及相对温和的压力下,很难达到美国能源部提出的2015年储氢标准(5.5%).10
为了进一步提高储氢量,近来又提出了一种在已有材料中掺杂金属Li、Mg、Ca等来提高材料储氢性能的方法.11-13Yang等13的研究表明,在掺杂不同金属的沸石材料中,Li掺杂的低硅铝X型沸石分子筛在77 K、0.1 MPa下储氢量可以提高到1.5%,Ca掺杂的低硅铝X型沸石分子筛在298 K、10 MPa下储氢量可以达到0.50%,为该类材料中最高,但这与2015年的储氢标准相差甚远.此外,这种掺杂金属的方法在实验操作上难度很大,而且通过合成、提纯,检测储氢能力是一个费时的过程,用实验方法寻找设计新的满足应用条件的储氢材料是极具有挑战性的工作.
利用分子模拟方法预测材料的储氢性能可以为实验设计新的储氢材料提供指导和帮助,降低研发周期和成本.分子模拟中的一个关键因素是分子力学力场(以下简称力场).通用力场例如UFF不能很好地体现骨架与氢气分子之间的相互作用.5,14,15而通过拟合实验数据得到的力场因受到实验数据精确性的限制不能保证参数的可靠性.16这些问题限制了分子模拟方法准确预测新材料性质的能力.目前,用量子化学计算已能精确地描述小分子间的相互作用.由于计算本身不依赖实验数据,因而被称为第一性原理计算.然而,对于含有大量粒子的化学平衡问题又必须用统计力学方法描述,因此,基于量子化学数据推导的力场是连接量子化学和统计力学计算的桥梁.17,18
本文以PAF材料为基本模型,设计了一个包含二价金属阳离子(Mg2+、Ca2+)及羧酸根的吡啶-2,6-二羧酸盐有机金属官能团作为构造单元修饰PAF.引入这样的官能团可以调节材料的吸附表面积、空腔体积以及材料表面与氢分子的结合能,从而提高PAF材料的储氢性能.我们运用量子化学方法计算确定了这些官能团和氢分子的相互作用,拟合量子化学计算数据建立了描述氢分子和新构建的材料表面相互作用的力场,进而利用巨正则系综蒙特卡洛(GCMC)模拟预测得到该种新型材料的平衡储氢性能.
2 计算模型和方法
2.1 计算模型
修饰到PAF上的是吡啶-2,6-二羧酸盐官能团(C7H3NO4M,M=Mg,Ca),如图1所示.PAF-3、PAF-4的材料空腔很大,骨架上有很多位点可以用来修饰该官能团,我们选择在每个骨架的有机连接体上修饰两个这种官能团(图2),以增大材料的吸附表面积.对于PAF-3,有两种方法修饰:PAF-3-dcM(type 1),PAF-3-dcM(type 2),如图2所示.“dc”表示双羧基,“M”表示金属镁或钙,下文皆用此命名.对于PAF-4,修饰方法有很多,但是经测试发现几种修饰后PAF-4的表面积和自由体积差别很小,因此只选择其中一种PAF-4-dcM做吸附预测比较.晶体结构详细参数见表1.
2.2 量子化学计算
量子化学计算方法使用MP2方法及TZVPP基组优化结构,用MP2方法、QZVPP基组加基组重叠误差(BSSE)校正计算能量.根据我们18之前的工作,用这样的方法得到的氢分子与含有金属镁、钙的有机基团的结合能数据接近包含电子相关效应的CCSD(T)及完全集组(CBS)计算的结果.我们首先对吡啶-2,6-二羧酸盐(C7H3NO4M,M=Mg,Ca)结构进行优化,得到的优化结构用来扫描和氢分子的相互作用势能面.扫描过程中金属羧酸配合物固定为优化结构,氢分子键长固定为0.74 nm,仅计算BSSE校正的单点能.H2分子与金属羧酸配合物的结合能按照如下公式计算:
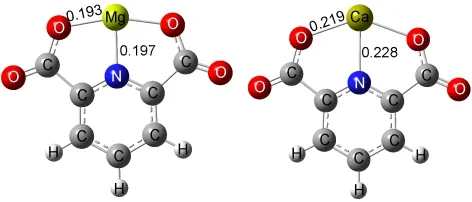
图1 吡啶-2,6-二羧酸镁、钙的优化结构Fig.1 Optimized structures of the functional group pyridine-2,6-dicarboxylate C7H3NO4M(M=Mg,Ca)bond length in nm
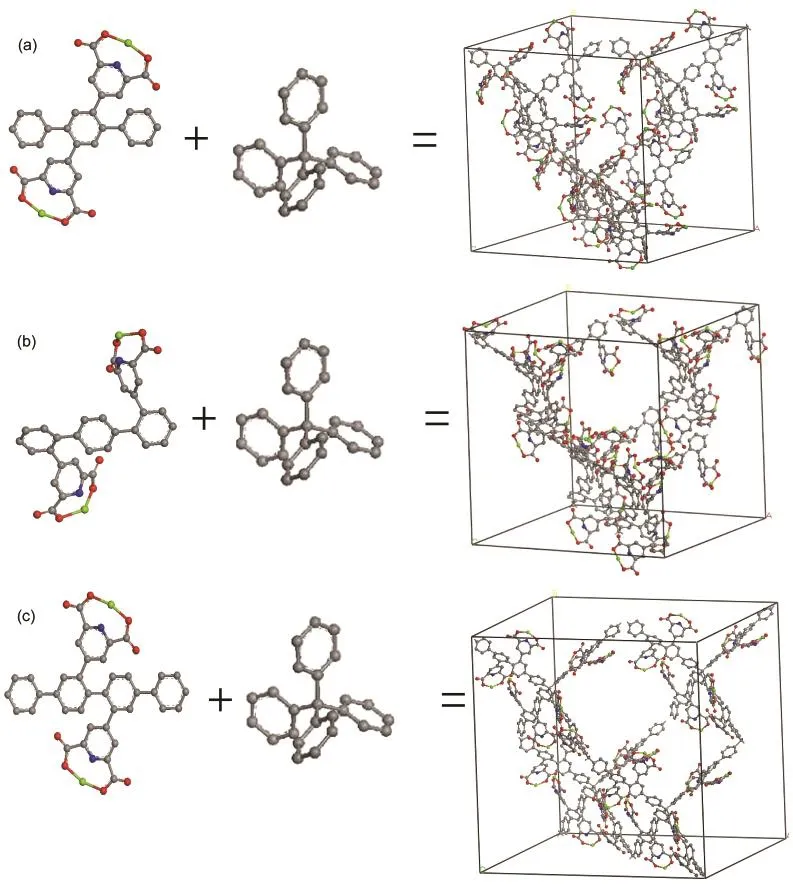
图2 PAF-3-dcM及PAF-4-dcM的搭建方式示意图Fig.2 Demonstration of different ways to build PAF-3-dcM and PAF-4-dcM (a)PAF-3-dcM(type 1),(b)PAF-3-dcM(type 2),(c)PAF-4-dcM;dc:two carboxyls,M=Mg,Ca
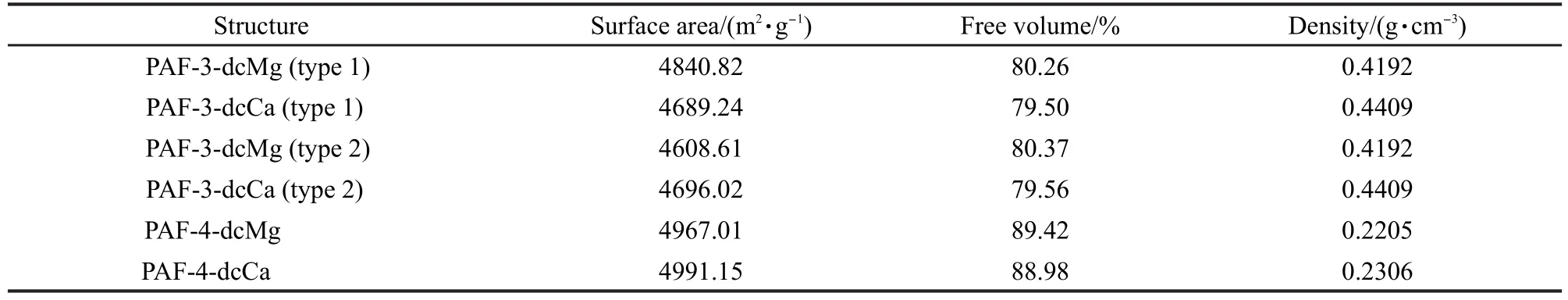
表1 晶体的自由体积、Langmuir表面积和密度结构数据Table 1 Free volumes,Langmuir surface areas,and density structure data of crystals
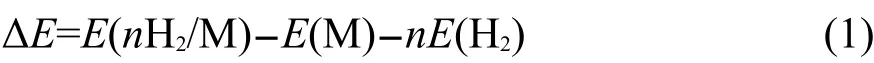
其中,ΔE为结合能,E(M)为金属羧酸配合物的能量,E(H2)为氢分子的能量,E(nH2/M)为氢分子与金属羧酸配合物结合后体系的能量,n为氢分子数.
拟合力场参数所需要的原子电荷用密度泛函理论(DFT)19方法计算的分子静电势(ESP)得到.DFT计算使用Gaussian 03软件包,20MP2计算使用TURBOMOLE 6.2软件包.21
2.3 力 场
分子模拟采用固定晶体内坐标的刚性骨架模拟,即不考虑分子内的运动.分子间的非键相互作用由原子对(pair-wise)之间的电荷(columbic)以及范德华(vdW)势函数描述.范德华势采用Lennard-Jones(LJ)12-6函数形式:
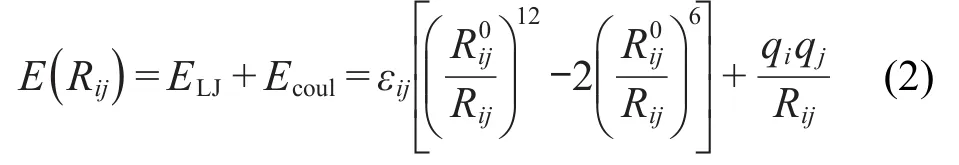
其中,Rij为i,j两个相互作用原子之间的距离,R0ij为平衡距离,此时的两个原子间相互作用能最大,为此时的曲线势能面的深度,也就是通常所说的势阱.式中的原子电荷(q)通过力场拟合DFT计算的静电势得到.不同原子间的Lennard-Jones参数采用Lorentz-Berthlot混合规则得到:
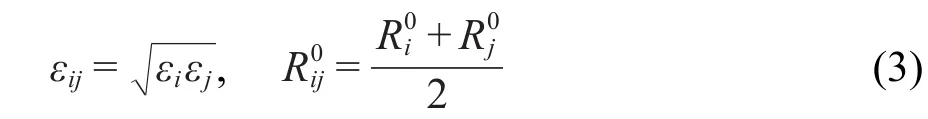
氢分子的势函数采用Darkrim以及Levesque提出的模型,18即氢分子键长固定为0.0741 nm.范德华作用位点位于分子的质心,参数ri=0.332 nm,εi= 0.305 kJ·mol-1.两个氢原子以及质心处放置重现氢分子四极矩的电荷,其中氢原子上电荷为0.4664e,质心处电荷为-0.9328e,分子保持电中性.
2.4 量子效应校正
氢分子质量很小,当温度很低(77 K)时,氢分子表现出明显的波动性,必须考虑量子效应的影响.14本工作中采用Feynman-Hibbs(FH)相互作用势函数反映量子效应.22,23通过拟合FH势函数得到LJ 12-6的有效势函数进行蒙特卡洛模拟.这个校正仅对范德华作用项,而不牵涉到电荷参数,其表达式为:

其中μ是一对相互作用原子的约化质量;U(r)是经典力场的相互作用势能,即LJ 12-6相互作用势能项, Uʹ(r)、Uʹ(r)、Uʹʹ(r)、Uʹʹ(r)分别是经典作用势能项对于相应的原子对作用距离的一阶、二阶、三阶、四阶导数.
2.5 巨正则系综蒙特卡洛模拟
采用巨正则系综蒙特卡洛模拟方法预测氢吸附的等温线.GCMC需要的化学势用等温等压系综(NPT)蒙特卡洛模拟及Widom粒子插入法24在指定温度,压力条件下得到.模拟采用TOWHEE-4.16.8程序,25模拟的细节如前报道.26GCMC中的蒙特卡洛移动包括粒子插入删除、平动以及转动,其对应的接受概率分别为30%、40%以及30%.运行1.0×106移动作为预平衡,接着运行1.0×106移动收集数据.模拟中范德华作用截断半径为1.3 nm,静电作用采用Ewald方法进行计算.
模拟得到的吸附量以两种形式表达:绝对质量吸附量以及超额质量吸附量.根据美国能源部最新公布的计算标准,绝对质量吸附量的计算公式如下:

其中Nads是吸附剂中吸附氢气分子的总数量,MH2是氢气的摩尔质量,MPAF是PAF材料的摩尔质量.实验数据一般以超额吸附量报道.超额吸附量用下式计算:
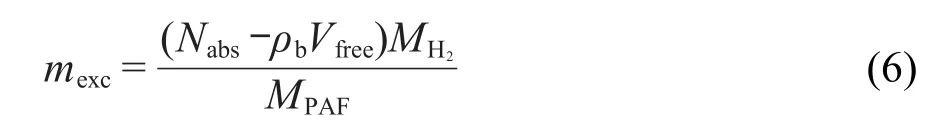
其中ρb为该温度压力下氢气的数密度,Vfree是PAF材料的自由体积,用Material Studio程序27计算.
体积吸附量公式定义如下:

其中VPAF代表PAF材料的总体积.
3 结果与讨论
3.1 量子化学计算
优化氢分子与含有金属镁、钙的金属羧酸配合物的团簇结构时,氢分子被逐个加入.首先选择靠近阳离子的位置,当阳离子周围位点饱和后,重复优化过程在氧原子周围逐个加入氢分子优化结构并计算单点能.从量子化学结构优化结果来看(图3), Mg2+周围有3个吸附位点,Ca2+有6个吸附位点,但其中4个位点与O原子共用.平均每个O原子周围有2个吸附位点.Mg2+与氢分子的距离在0.22到0.23 nm左右,Ca2+与氢分子距离在0.27到0.28 nm左右.总体来说,每个吡啶-2,6-二羧酸镁、钙配合物可以分别提供13、14个氢分子吸附位点.

图3 C7H3NO4M(M=Mg,Ca)氢气吸附的优化结构图Fig.3 Optimized structures of C7H3NO4M(M=Mg,Ca) adsorption with hydrogen moleculesCa2+shared 4 hydrogen molecules with O sites nearby.distance in nm
计算得到的氢分子与含有金属镁、钙的金属羧酸配合物的总结合能和平均结合能如图4所示.在氢分子刚加入的初期,氢分子与Mg2+的结合能是-24.3 kJ·mol-1,与Ca2+的结合能为-14.8 kJ· mol-1.Mg2+因为与氢分子的距离更近(约0.22 nm),所以比Ca2+与氢分子(约0.27 nm,图3)的结合更强.随着氢分子的逐个加入,阳离子周围的吸附位点被占满后,其余的氢分子倾向于分布在羧基中的氧原子周围,这时Ca2+与氢分子的平均结合能(-8.72 kJ· mol-1)变得更负,小于Mg2+与氢分子的平均结合能(-7.71 kJ·mol-1).出现这种反超现象主要是因为Ca2+的离子半径(0.219 nm)比Mg2+(0.193 nm)大,与氢分子的接触面积更大.从总结合能的角度可以更清晰地看到这一反超的现象.总体来说,当阳离子和羧基的吸附位点都被占满后,官能团与氢分子的平均结合能在-8 kJ·mol-1左右,结合能大小对于吸附和脱附过程都很合适,方便未来车载储氢的实际应用.之前文献28中报道的储氢材料最优吸附热在15 kJ·mol-1左右,主要是基于碳硅纳米管类材料,我们所提出的新型离子型官能团能够提供更多的吸附位点,在下面的讨论中可以看出没有必要要求过高的吸附能.
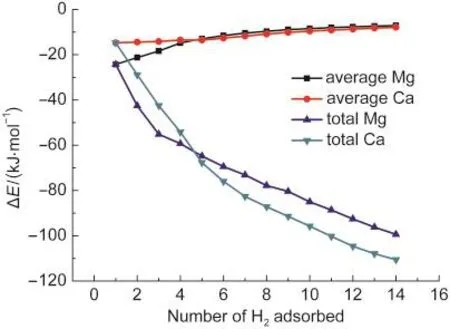
图4 C7H3NO4M(M=Mg,Ca)官能团与氢分子的总结合能和平均结合能(ΔE)Fig.4 Total and average binding energies(ΔE)between the functional group C7H3NO4M(M=Mg,Ca)and the hydrogen moleculesBSSE corrections were included at the MP2/QZVPP level of theory.
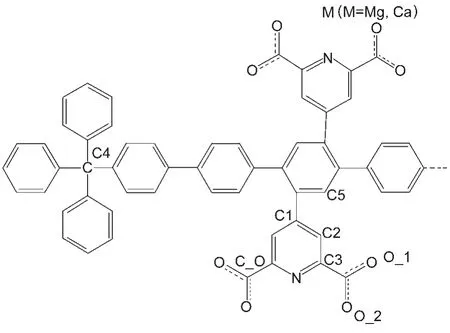
图5 原子类型的定义Fig.5 Demonstration of atom types
3.2 力场参数化
根据官能团在PAF中的不同化学环境我们定义了12种不同的原子类型(图5).对于每种原子类型的参数,首先计算两种不同金属羧酸配合物的ESP电荷和静电势,然后拟合量化的静电势,并利用键增量(bond-increment)方案得到不同原子类型之间的电荷转移值,从而最后得到每个原子上所带的电荷.整个过程用DFF软件包自动完成.
由于模拟中骨架是固定的,所以我们只拟合非键参数.固定金属羧酸配合物和H2分子的键长、键角和二面角值.拟合的数据是量子化学计算的H2分子与不同金属羧酸配合物相互作用的结合能.通过调整范德华参数,使力场计算的H2分子与金属羧酸配合物相互作用的结合能与量子化学计算值相吻合.
力场优化的方案如下:首先以量子化学计算得到的含H2分子的金属羧酸配合物的结构为目标优化非键参数中的vdW直径.然后综合调整vdW直径和阱深拟合能量部分.与量子化学计算结果进行对比,如果吻合,则参数是合理的,不用再调整,如果差距较大,则需要继续调整参数.最后我们得到的力场参数(表2)可以较为合理地预测H2与金属羧酸配合物的相互作用(表3).
3.3 PAF-3-dcM,PAF-4-dcM(M=Mg,Ca)的氢吸附能力
3.3.1 绝对吸附量
图6是模拟得到的在77和233 K下,压力从0到30 MPa的PAF-3-dcM(type 1)和PAF-4-dcM(M= Mg,Ca)的氢气绝对质量吸附等温线.由于PAF-3-dcM(type 2)的绝对质量吸附量和体积吸附量与type 1基本相同,未在图中列出.在77 K和30 MPa下,PAF-3-dcMg和PAF-3-dcCa绝对质量吸附量分别达到0.180和0.169 kg·kg-1.当温度升高到233 K, PAF-3-dcMg和PAF-3-dcCa绝对质量吸附量依然能够达到0.071和0.068 kg·kg-1,超过了美国能源部提出的2015年储氢目标(0.055 kg·kg-1,即5.5%).由于PAF-4-dcM的密度要远小于PAF-3-dcM,所以它的绝对质量吸附量结果更好.在77 K、30 MPa下, PAF-4-dcMg和PAF-4-dcCa绝对质量吸附量分别达到0.351和0.337 kg·kg-1.当温度升高到233 K, PAF-4-dcMg和PAF-4-dcCa绝对质量吸附量依然能够达到0.131和0.125 kg·kg-1,远远超过了美国能源部提出的2015年储氢目标.
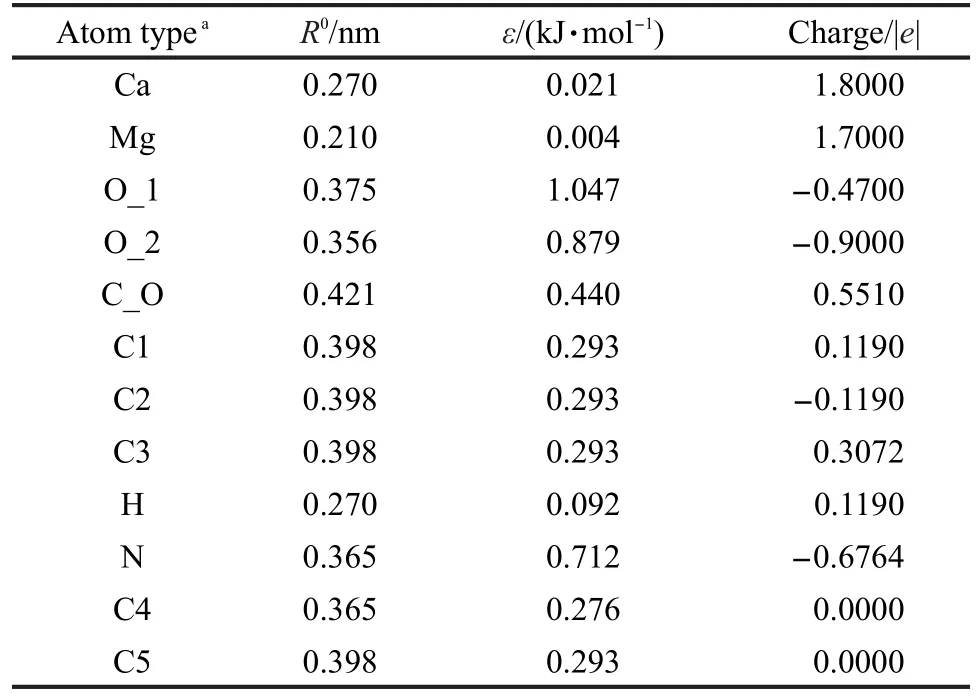
表2 原子类型、非键参数(LJ 12-6)和电荷参数Table 2 Atom types,associated nonbond(LJ 12-6)and charge parameters
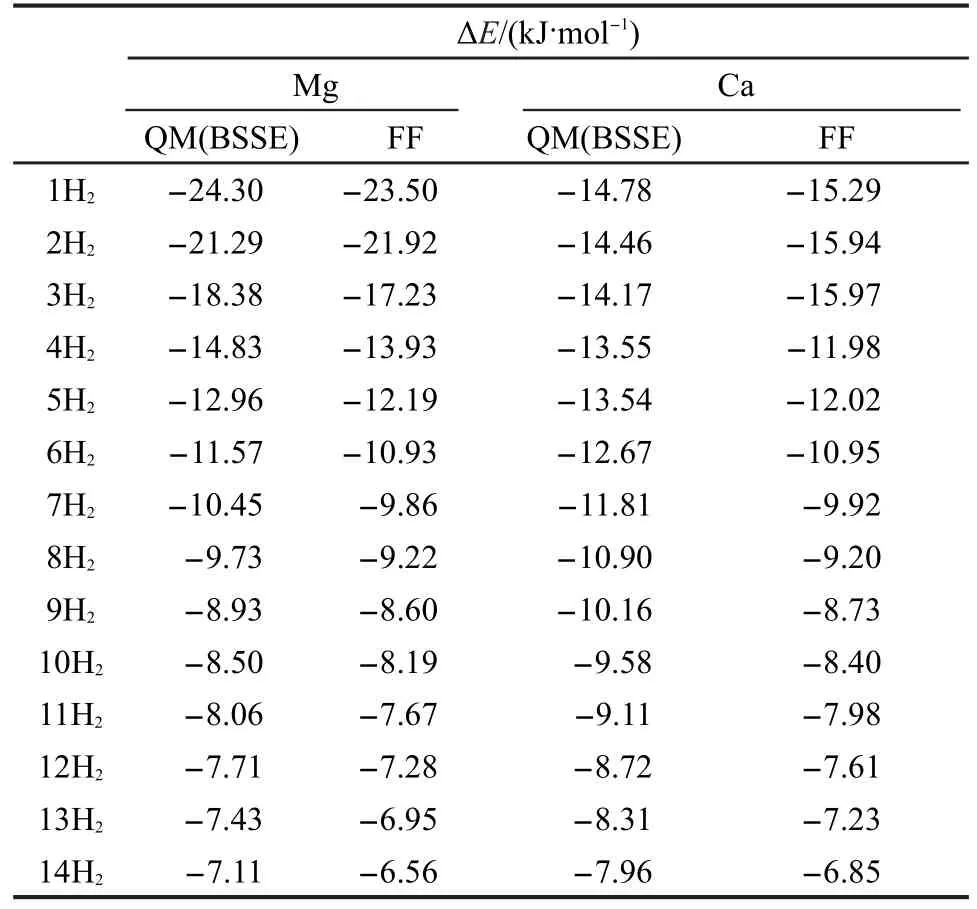
表3 量子化学和力场计算的H2分子与不同C7H3NO4M (M=Mg,Ca)相互作用的平均结合能对比Table 3 Comparison of the average binding energies of H2 on C7H3NO4M(M=Mg,Ca)between quantum mechanics (QM)and force field(FF)calculations
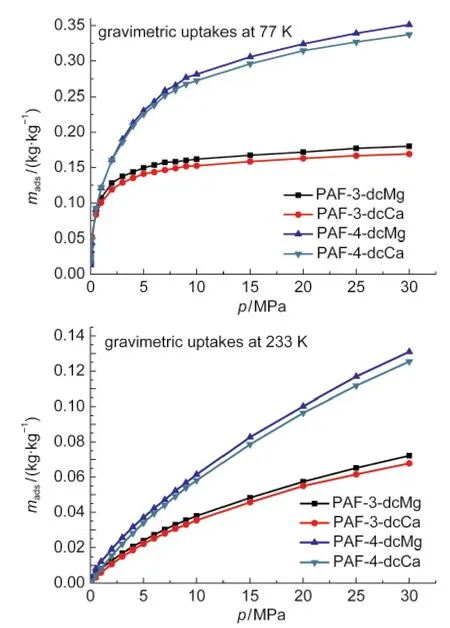
图6 PAF-3-dcM(type 1)和PAF-4-dcM(M=Mg,Ca)的绝对质量吸附量(mads)Fig.6 Absolute gravimetric adsorption(mads)curves ofPAF-3-dcM(type 1)and PAF-4-dcM(M=Mg,Ca)
图7是和图6相对应的绝对体积吸附量.因为PAF-3-dcM总体积比PAF-4-dcM小很多,因此在77 K和低压条件下,PAF-3-dcM的体积吸附量大于PAF-4-dcM.但是当压力提高到30 MPa,PAF-4-dcM因为自由体积和表面积更大而在高压时体积吸附量反超了PAF-3-dcM.当温度升高到233 K,氢分子在吸附位点振动加剧,吸附表面积、自由体积等因素对吸附结果影响减弱,材料的总体积成为影响体积吸附量大小的决定因素,因此,在233 K条件下, PAF-3-dcM的体积吸附量普遍高于PAF-4-dcM.在77 K、30 MPa下,PAF-3-dcMg和PAF-3-dcCa绝对体积吸附量分别达到75.5和74.6 g·L-1.当温度升高到233 K,PAF-3-dcMg和PAF-3-dcCa绝对体积吸附量依然能够达到30.3和29.9 g·L-1.在77 K、30 MPa下,PAF-4-dcMg和PAF-4-dcCa绝对体积吸附量分别达到77.4和77.7 g·L-1.当温度升高到233 K, PAF-4-dcMg和PAF-4-dcCa绝对体积吸附量能够达到28.9和28.9 g·L-1.

图7 PAF-3-dcM(type 1)和PAF-4-dcM(M=Mg,Ca)的绝对体积吸附量(Vads)Fig.7 Absolute volumetric adsorption(Vads)curves of PAF-3-dcM(type 1)and PAF-4-dcM(M=Mg,Ca)
3.3.2 超额吸附量
超额吸附量更好地显示了材料对吸附的贡献.图8是模拟得到的PAF-3-dcM(type 1和type 2),以及未修饰的PAF-3的超额质量吸附量.从中可以清楚地发现,通过与未进行任何修饰的纯PAF材料氢气吸附量对比,引入的新型官能团可以有效提高PAF的储氢量.PAF-3-dcMg(type 1)的超额吸附量在同类材料中最高,这与其自身吸附表面积最大(4840.82 m2·g-1),密度最小(0.4192 g·cm-3)的结构特点相一致.PAF-3-dcCa(type 1)和PAF-3-dcCa(type 2)晶体参数基本相同,所以这两种材料的吸附表现也基本相同,但因这两种材料密度稍大使得它们的吸附结果略低于PAF-3-dcMg(type 1).值得一提的是,PAF-3-dcMg(type 2)的吸附表现比以上三种材料都要差,在材料自由体积、密度基本相同的条件下,因为PAF-3-dcMg(type 2)的吸附表面积比type 1下降很多,为此类材料中最低(4608.61 m2·g-1),导致它的吸附结果低于另外三种材料.由此可见,在其他结构特征基本相同的条件下,储氢材料的吸附表面积下降会直接导致氢气吸附量的降低.

图8 PAF-3-dcM(type 1,type 2)以及纯PAF-3的超额质量吸附量(mexc)Fig.8 Excess gravimetric adsorption(mexc)curves of PAF-3-dcM(type 1,type 2)and pure PAF-3
4 结论
用MP2方法,TZVPP基组以及BSSE校正计算了氢分子与修饰在PAF骨架上的羧酸镁、羧酸钙官能团的相互作用,并建立了描述这一相互作用的分子力学力场.在此基础上用巨正则系综蒙特卡洛模拟预测了氢气在该种新型PAF材料上的吸附等温线.
用量子化学计算得到大量的能量数据,可以从势能面的角度客观地确定力场参数.量子化学计算结果表明,每个羧酸镁、羧酸钙官能团分别可以提供13、14个氢分子吸附位点,与每个氢分子的平均结合能在-8 kJ·mol-1左右.根据官能团在PAF中的不同化学环境我们定义了12种不同的原子类型,通过调整原子电荷参数和范德华参数,使力场计算的H2分子与金属羧酸配合物相互作用的结合能与量子化学计算值相吻合,从而得到一套可靠的力场参数用于模拟预测氢气吸附.
通过比较不同温度和压力下材料的绝对吸附量和超额吸附量可以看出,在PAF中引入羧酸镁、羧酸钙官能团可以显著提高材料的综合储氢性能,达到并超过了美国能源部提出的2015年储氢标准.同时该工作还揭示了氢气吸附量与材料的表面积,空腔体积和分子作用强度间的复杂关系.一般来说,在低负载时表面积是决定吸附量的主要因素,在高负载时空间体积是主要因素.分子作用强度直接影响吸附量,但在高温时其作用并不明显.
(1) Millward,A.R.;Yaghi,O.M.J.Am.Chem.Soc.2007,127, 17998.
(2)Wong-Foy,A.G.;Matzger,A.J.;Yaghi,O.M.J.Am.Chem. Soc.2006,128,3494.
(3) Kaye,S.S.;Dailly,A.;Yaghi,O.M.;Long,J.R.J.Am.Chem. Soc.2007,129,14176.
(4) Han,S.S.;Deng,W.Q.;Goddard,W.A.Angew.Chem.Int. Edit.2007,46,6289.
(5) Frost,H.;Duren,T.;Snuff,R.Q.J.Phys.Chem.B 2006,110, 9565.
(6) Zeng,Y.Y.;Zhang,B.J.Acta Phys.-Chim.Sin.2008,24,1493. [曾余瑶,张秉坚.物理化学学报,2008,24,1493.]
(7) Mu,H.;Liu,D.H.;Yang,Q.Y.;Zhong,C.L.Acta Phys.-Chim. Sin.2010,26,1657.[穆 韡,刘大欢,阳庆元,仲崇立.物理化学学报,2010,26,1657.]
(8) Farha,O.K.;Yazaydın,A.;Eryazici,I.;Malliakas,C.D.; Hauser,B.G.;Kanatzidis,M.G.;Nguyen,S.T.;Snurr,R.Q.; Hupp,J.T.Nat.Chem.2010,2,944.
(9) Ben,T.;Ren,H.;Ma,S.Q.;Cao,D.P.;Lan,J.H.;Jing,X.F.; Wang,W.C.;Xu,J.;Deng,F.;Simmons,J.M.Angew.Chem. Int.Edit.2009,48,9457.
(10) U.S.Department of Energy.Energy Efficiency and Renewable Energy.http://www1.eere.energy.gov/hydrogenandfuelcells/ storage/pdfs/targets_onboard_hydro_storage.pdf.
(11) Sun,Y.X.;Ben,T.;Wang,L.;Qiu,S.L.;Sun,H.J.Phys.Chem. Lett.2010,1,2753.
(12)Yoon,M.;Yang,S.;Hicke,C.;Wang,E.;Geohegan,D.;Zhang, Z.Phys.Rev.Lett.2008,100,206806.
(13) Wang,L.F.;Yang,R.T.Ind.Eng.Chem.Res.2010,49,3634.
(14) Garbemglio,G.;Skoulidas,M.;Johnson,J.K.J.Phys.Chem.B 2005,109,13094.
(15) Frost,H.;Snurr,R.Q.J.Phys.Chem.C 2007,111,18794.
(16)Yang,Q.;Zhong,C.J.Phys.Chem.B 2005,109,11862.
(17) Fu,J.;Sun,H.J.Phys.Chem.C 2009,113,21815.
(18) Wang,L.;Sun,Y.X.;Sun,H.Faraday Discuss.2011,151,143.
(19) Heinz,H.;Suter,U.W.J.Phys.Chem.B 2004,108,18341.
(20) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision C.02;Gaussian Inc.:Wallingford,CT,2004.
(21)Ahlrichs,R.;Bär,M.;Häser,M.;Horn,H.;Kölmel,C.Chem. Phys.Lett.1989,162,165.
(22)Feynman,R.P.;Hibbs,A.R.Quantum Mechanics and Path Integrals;McGraw-Hill:New York,1965.
(23) Feynman,R.P.Mod.Phys.1948,20,367.
(24) Allen,M.P.;Tildesley,D.J.Computer Simulation of Liquids; Oxford University Press:New York,1987.
(25)Martin,M.G.MCCCS Towhee,2006,http://towhee.sourceforge. net/.
(26) Liu,L.C.;Fu,J.;Sun,H.Science in China Series B:Chemistry 2008,38,331.[刘连池,付 嘉,孙 淮.中国科学B辑:化学,2008,38,331.]
(27) Materials Studio;Accelrys Inc.:San Diego,CA.
(28) Bhatia,S.K.;Myers,A.L.Langmuir 2006,22,1688.
November 14,2011;Revised:December 26,2011;Published on Web:December 30,2011.
Predicting Hydrogen Storage Performances in Porous Aromatic Frameworks Containing Carboxylate Functional Groups with Divalent Metallic Cations
MIAO Yan-Lin SUN Huai*WANG Lin SUN Ying-Xin (College of Chemistry and Chemical Engineering,Shanghai Jiao Tong University,Shanghai 200240,P.R.China)
We report force field predictions for the hydrogen uptakes of porous aromatic framework(PAF) materials containing carboxylate functional groups with divalent metallic cations.The ab initio calculations were performed on our proposed functional groups and hydrogen molecules using the MP2 method with the TZVPP basis set and basis set superposition error(BSSE)correction.A force field was developed based on the ab initio energetic data.The resulting force field was applied to predict hydrogen adsorption isotherms at different temperatures and pressures using the grand canonical Monte Carlo(GCMC) method.Each functional group of divalent metallic cations and two carboxylic acid groups provided 13(Mg) or 14(Ca)binding sites for hydrogen molecules with an average binding energy of 8 kJ·mol-1per hydrogen molecule.The predicted hydrogen adsorption results were improved remarkably by the functional groups at normal ambient conditions,exceeding the 2015 target set by the department of energy(DOE)of USA.This work reveals the complex relationship between hydrogen uptake and surface area,and the free volumes and binding energies of different materials.
Hydrogen storage;Dicarboxylate;Porous aromatic framework;Ab initio;Molecular simulation
10.3866/PKU.WHXB201112301
O642
∗Corresponding author.Email:huaisun@sjtu.edu.cn;Tel:+86-21-54748987-601.
The project was supported by the National Natural Science Foundation of China(21073119)and National Key Basic Research Program of China (973)(2007CB209701).
国家自然科学基金(21073119)及国家重点基础研究发展计划(973)(2007CB209701)资助
猜你喜欢
航空材料学报(2023年6期)2023-12-18 05:23:50
小学生学习指导(小军迷联盟)(2023年3期)2023-03-27 09:22:44
大学物理(2022年9期)2022-09-28 01:10:52
中国特种设备安全(2022年4期)2022-07-08 02:41:40
中国特种设备安全(2022年4期)2022-07-08 02:41:28
中国音乐学(2022年1期)2022-05-05 06:48:46
物理通报(2020年7期)2020-07-01 09:28:02
物理学报(2018年10期)2018-06-14 06:31:32
信息记录材料(2016年4期)2016-03-11 15:22:31
材料科学与工程学报(2016年5期)2016-02-27 07:11:37
