
Alexander病 1例报告与文献复习
2010-08-25 03:55:28杨任民周志华叶群荣李清清胡文彬
中风与神经疾病杂志 2010年1期
杨任民, 周志华, 叶群荣, 李清清, 胡文彬
1949年 Alexander尸检 1例于发病后 8个月内死亡的15个月婴儿,临床表现为智能、运动发育迟滞及脑积水,病理发现于大脑白质及软膜下广泛地被一种称做 Rosenthal纤维分布在星状胶质细胞内,故命名为进行性白质变性症。此后进一步证实患者神经纤维酸蛋白(glial fibrillary acidic protein,GFAP)存在位于 17q21的胶质基因突变,引起星状胶质细胞内 GFAP和热休克蛋白(α,β-Crystallin,heat shock protein-27)过度蓄积,造成星形胶质细胞功能障碍所致白质广泛脱髓鞘变性[1,2];并根据临床、影像学和病理特征被普遍承认为属常染色体显性遗传的独立的疾病[3,4],并根据不同的发病年龄和不同的临床表现,可分为婴儿型、少年型和成人型[5,6]。过去本病都依靠病理诊断,自影像学广泛应用以来,MRI对诊断本病具有重要价值[7~9]。不少患者因此获得生前早期诊断。目前全世界约有近百篇报道,但国内文献仅有 3篇 4例个案报道,均为病理诊断,其中 2例为婴儿型[10],另 2例为成人型[11,12]。作者等最近遇见 1例临床与影像学较为典型的少年型Alexander病,报道如下。
患儿,女,2岁 6个月。发现其进行性右侧上下肢持物无力、头颈部后仰、言语困难 1个月余于 2009年 2月 17日入院。患儿系 2006年 9月 1日足月剖宫产出生,于出生 1周岁已能独立行走和会叫“爸、妈”。在 2009年 1月初,患儿亲属发现其行走时右下肢无力,易摔跤,并随意抓周围物品啃咬,但未予重视。1月中旬,发现其出现持续性右上肢不自主后旋,头颈部后仰伴挺胸、抬腹等不自主运动,尤于情绪激动时明显,睡眠时消失;并语速明显缓慢,语词较前明显贫乏,但口齿尚较清楚,上述症状呈进行性加重。1月下旬,右侧上下肢无力,持物及行走不能,并仅能说“爸爸、妈妈、哥哥”等单字,曾至省内外各大医院就诊,被诊断为“病毒性脑炎”,予“营养神经、活化脑代谢、降颅压、甲强龙 0.25g冲击治疗及大剂量丙种球蛋白静脉注射等治疗”,症状无改善并渐加重,出现言语不能、吞咽困难,不自主张口、吐舌等不自主运动。2月初,患儿右上肢出现不自主抖动,频繁啃咬自己双手。病程中,患儿无意识丧失、发作性全身抽搐及呕吐等。睡眠尚可。否认有一氧化碳中毒史,否认食用霉甘蔗或服用山豆根等中药史。按时接种疫苗,无不良反应。父母非近亲婚配。家族中无类似病患者。
入院查体:神清,查体欠合作,言语不能,吞咽困难。可见头颈部持续性不自主后仰,及张口、吐舌、挺胸、抬腹样等不自主动作。颈稍抵抗,克、布氏征(-)。左侧上下肢肌力正常,肌张力呈轻度折刀样增高,右上肢向后扭转,右侧上下肢肌力Ⅳ°,肌张力右上肢呈轻度铅管样增高,右下肢减低;四肢腱反射(+),双侧 Babinski征(-),左手强握反射(+)。右手可见轻度阵挛。双手可见多处咬痕。
实验室及特殊检查:血清单纯疱疹病毒 1型、2型抗体IgG均阳性,风疹病毒抗体 IgG、IgM均阳性,巨细胞病毒抗体IgG阳性。MRI(Flair WI)示额顶部较后枕部信号低,双侧苍白球、壳核、尾状核对称性明显肿胀,压迫侧脑室,使侧脑室变狭及胼胝体明显变薄;侧脑室前角“周围边样变”(periventricular rim),中脑、延脑可见斑片状长 T2异常信号(见图1~图4)。血常规正常,棘红细胞计数:1‰ ~3‰,电解质、肝肾功能、铜生化、血免疫球蛋白、血乳酸最小运动量试验等均正常,EEG示正常范围脑电图,心电图正常。
诊断:Alexander病。
讨 论
Alexander病根据发病年龄不同,临床表现与影像学所见也迥异,一般分为婴儿型、少年型及成人型 3型[11],近年更在婴儿型中提出新生儿型亚型[13,14]。(1)婴儿型[15~16]:是Alexander病中发病率最高的一个类型,占本病总数的约80%。大多在出生后至 2岁内(平均 6个月)发病,男性多见。从婴儿早期便表现精神运动发育迟滞,由于 Rosenthal纤维往往阻塞导水管而发生脑积水,故出生后头围已较大,并逐渐缓慢增大,至出生后 6~18个月,便超过正常婴儿近一倍。癫痫发作常为本型的初发症状,可发生癫痫持续状态和婴儿痉挛,且往往属难治性癫痫;此外尚有肌张力减低或痉挛性增高,吸吮困难、构音障碍等真性或假性延髓麻痹。常在 10岁前死亡。(2)少年型[15~16]:发病年龄从 2岁至 20岁,临床表现与婴儿型颇多相似处,尤以真性、假性延髓麻痹为特征。但运动功能退行较轻,癫痫发作较少见;智能退行较轻且较缓慢,也有智能障碍不明显者;巨头症也不显著,头围往往正常。(3)新生儿型[14]:是婴儿型中具有特殊临床特征的亚型,于出生后 1月以内大多以明显的脑积水和颅压增高发病,出生后精神运动方面几乎不发育或急速退行,延髓麻痹为其主征,缓慢进行性加重。脑脊液蛋白增高。(4)成人型[16~17]较少见。一般于 20~70岁发病,临床表现与少年型相似,以真性、假性延髓麻痹为主征,往往出现软腭及全身肌阵挛。Li等报道 44例 Alexander病的临床特征,其中 15例少年型的主要症状和发生率为[18]:(1)发病年龄 2.0~11.5岁,(2)假性/真性延髓麻痹 11/15,(3)认知功能障碍9/15,(4)肌阵挛 8/15,(5)共济失调 7/15,(6)癫痫发作 4/15,(7)巨颅 3/15,(8)平均生存 8.1年。
头部MRI是本病生前早期诊断的重要根据,Knaap等提出诊断标准[8]为下列 5项中具备 4项可诊断为 Alexander病:(1)额部为主的广泛脑白质异常信号;(2)出现脑室周围边;(3)基底节和丘脑明显肿胀(T2WI呈高信号);(4)脑干有异常信号;(5)在脑室周围、额叶白质、基底节、丘脑、视交叉、穹窿、小脑齿状核及脑干有增强效应。
本病例为 2岁 5个月发病,临床以进行性言语不清、吞咽困难为主征,并认知障碍,引起自咬手,右手轻度肌阵挛等。头部 MRI(Flair WI)可见额顶部较枕部信号为低;双侧苍白球、壳核及尾状核明显对称肿胀,压迫双侧侧脑室,在侧脑室前角有边样异常改变,脑干可见斑片状长 T2异常信号。因此,符合 Alexander病的影像学诊断。
本病无有效治疗方法,虽近年有报道磷酸肌醇-3-激酶和木斛皮素可能减少 GFAP表达,但尚处于动物试验阶段。
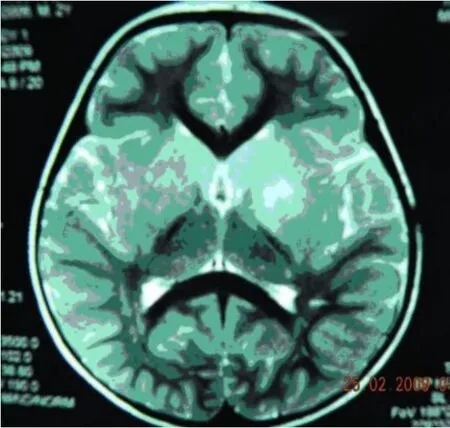
图1 双侧基底节区明显肿胀,侧脑室受压
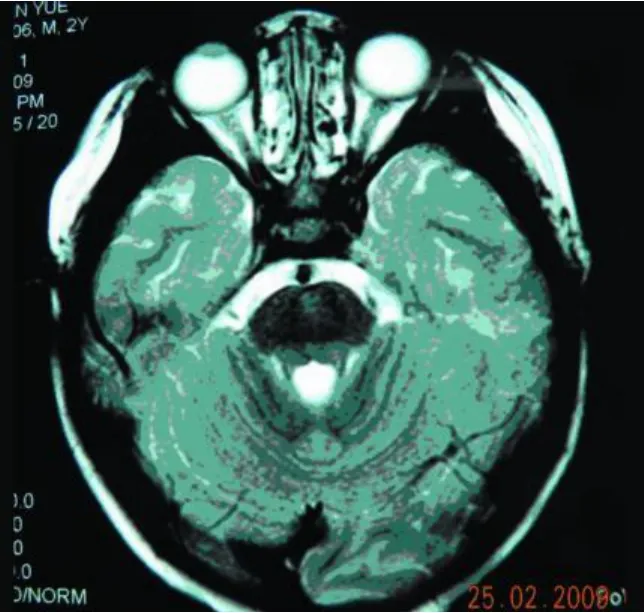
图2 脑干长 T2异常信号
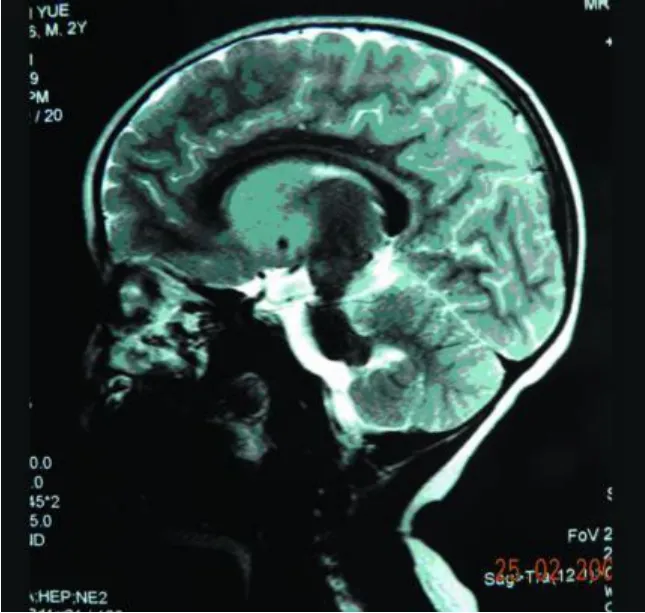
图3 大脑前半部较后半部信号偏低

图4 双侧脑室前角“周围边样”改变
[1] Alexander WS.Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant[J].Brain,1949,72:273-281.
[2] Brenner M,Johnson AB,Boespflug-Tanguy O,et al.Mutations in GFAP,encoding glial fibrillary acidic protein,are associated with Alexander disease[J].Nat Genet,2001,27(1):117-120.
[3] Namekawa M,Takiyama Y,Aoki Y,et al.Identification of GFAP gene mutation hereditary adult onset Alexander's disease[J].Ann Neurol,2002,52(6):779-785.
[4] Stumpf E,Masson H,Dequette A,et al.Adult Alexander disease with autosomal dominant transmission:a distinct entity caused by mutation in the glial fibrillary acidic protein gene[J].Arch Neurol,2003,60(9):1307-1312.
[5] Van der Knaap MS,Naidu S,Breiter SN,et al.Alexander disease:diagnosis with MR imaging[J].AJNR Am J Neuroradiol,2001,22(3):541-552.
[6] Russo LSJr,Aron A,Anderson PJ.Alexander's disease:areport and reappraisal[J].Neurology,1976,26(7):607-614.
[7] Schwankhaus JD,Parisi JE,Gulledge WR,et al.Hereditary adult-onset Alexander's disease with palatal myoclous,spastic paraparesis and cerebellar ataxia[J].Neurology,1995,45(12):2266-2271.
[8] Van Der Knaap MS,Ramesh V,Schittmann R,et al.Alexander disease:ventricular garlands and abnormalities of medulla and spinal cord[J].Neurology,2006,66(4):494-498.
[9] Farina L,Pareyson D,Minati L,et al.Can MR imaging diagnose adult-onset Alexander disease[J]?AJNRAm JNeuroradiol,2008,29(6):1190-1196.
[10] 朱杰明,鲍克容,郑英明,等.Alexander氏病 2例报告[J].临床儿科杂志,1993,11(1):34-35.
[11] 夏学锋,易咏红,廖已平,等.Alexander病的临床和病理研究[J].中华神经科杂志,1999,32:165-167.
[12] 胡 波,杨 欢,田晓琳,等.成人型 Alexander病 1例报告[J].临床神经病学杂志,2009,22:11.
[13] Jonnson AB,Brenner M.Alexander's disease:clinical,pathologic,and genetic features[J].J Child Neurol,2003,18:625-632.
[14] Spriger S,Erlewein R,Naegele T,et al.Alexander disease-classification revisited and isolation of a neonatal form[J].Neuropediatrics,2000,31(2):86-92.
[15] Sakuma H,Sasaki M.Clinical aspects of Alexander disease in Childhood[J].Neurol Med,2008,69:220-226.
[16] 张 艳,李存江.亚历山大病的诊断[J].中华神经科杂志,2007,40:709-711.
[17] Ohnari K,Tsuji S.An adult form of Alexander disease[J].Neurol Med,2008,69:227-231.
[18] Li R,Johnson AB,Salomons G,et al.Glial fibrillary acidic protein mutations in infantile,juvenile,and adult forms of Alexander disease[J].Ann Neurol,2005,57:310-326.
猜你喜欢
昆明医科大学学报(2021年4期)2021-07-23 01:21:46
甘肃科技(2020年20期)2020-04-13 00:30:54
中国心血管杂志(2020年4期)2020-01-09 03:47:19
中国学校体育(2018年5期)2018-05-14 15:46:17
医学信息(2018年5期)2018-04-20 11:03:20
新课程研究·教师教育(2017年3期)2017-05-26 11:20:50
中华骨与关节外科杂志(2017年1期)2017-05-17 06:11:27
临床与实验病理学杂志(2017年5期)2017-03-07 23:00:35
中国当代医药(2015年26期)2015-03-01 02:07:07
中国实用医药(2014年31期)2014-11-12 15:58:57
