
介导人α-反义寡核苷酸慢病毒载体的构建
2010-08-21 00:21赖颖晖赖永榕杨高晖
中国实验诊断学 2010年9期
赖颖晖,赖永榕,杨高晖
(广西医科大学第一附属医院血液科,广西南宁 530021)
介导人α-反义寡核苷酸慢病毒载体的构建
赖颖晖,赖永榕*,杨高晖
(广西医科大学第一附属医院血液科,广西南宁 530021)
目的构建介导人α-反义寡核苷酸的慢病毒载体,为α-反义寡核苷酸对β-地中海贫血基因治疗的体内试验提供稳定的转染细胞载体。方法根据人α-反义寡核苷酸序列设计siRNA、合成DNA片段,通过双限制性内切酶消化和连接的方法构建pGCSIL-vshR NA-GFP载体质粒,接着该质粒转化感受态的大肠杆菌E.coliDH5α,通过PCR及基因测序鉴定阳性克隆,再经Lipofectamine 2000将pGCSIL-vshRNA-GFP,pHelper 1.0和pHelper 2.0三质粒系统共转染293 T细胞包装病毒,通过绿色荧光蛋白的表达测定收集的病毒滴度。结果重组质粒的外源基因PCR鉴定正确,基因测序结果与所需要的α-反义寡核苷酸序列完全一致,浓缩后病毒滴度为5×109TU/ml。结论成功构建介导人α-反义寡核苷酸的慢病毒载体。
慢病毒载体;反义寡核苷酸;基因治疗
(Chin J Lab Diagn,2010,14:1358)
β-地中海贫血是一种对人类健康危害严重的遗 传性溶血性疾病。对重型β-地中海贫血,临床上缺乏有效的治疗手段,病人依赖于长期输血及去铁治疗来维持生命,费用巨大,死亡率极高[1]。造血干细胞移植是唯一的根治方法,但是由于合适供体来源
受限及费用昂贵,临床上难以广泛开展。基因治疗成为了β-地中海贫血治疗的希望。刘容容等[2]的研究所筛选出的反义寡核苷酸(ASON)能高效抑制人α-珠蛋白基因表达,并能有效地抑制体外培养的重型β-地中海贫血红系细胞α-珠蛋白基因表达,从而改善α/β+γ珠蛋白基因的比例失衡。本研究构建介导上述人α-ASON的慢病毒载体,并包装纯化慢病毒颗粒,为后续的α-ASON对β地中海贫血基因治疗的体内试验奠定基础。
1 材料与方法
1.1 主要试剂及仪器 病毒载体(pGC-LV重组载体,pHelper 1.0,pHelper 2.0)(上海吉凯基因技术有限公司);293T(上海吉凯基因技术有限公司);Age I和EcoRI(NEB);250bp DNA ladder Marker(捷瑞);Lipofectamine 2000(Invitrogen)。凝胶成像仪(天能公司),细菌摇床(华利达实验设备公司),细菌培养箱(上海一恒科学仪器有限公司),PCR仪(Applied Biosystems),荧光显微镜(奥林帕斯 micropublisher 3.3RTV),CO2培养箱(SANYO)。
1.2 实验方法
1.2.1 siRNA设计 根据刘容容等[3]筛选出的靶向人α-珠蛋白基因mRNA翻译起始区、能高效抑制α-珠蛋白基因表达的ASON 序列(5′-CAGCACCATGGTGGGTTCTC-3′),设计 siRNA 序列 为 CAGCACCATGGTGGGTTCTC。病毒载体构建框架见表1。

表1 病毒载体构建框架
1.2.2 合成DNA片段 合成片段信息:
PSCSI1907-1 CcggCAGCACCATGGTGGGTTCTCTTCAAGAGAGAGAACCCACCATGGTGCTGTTTTTg;PSCSI-1907-2 aattcaaaaaCAGCACCATGGTGGGTTCTCTCTCTTG AAGAGAACCCACCATGGTGCTG
委托上海吉凯基因技术有限公司合成引物。引物退火。
1.2.3 将α-ASON序列连接入慢病毒载体 Age I和EcoRI酶切pGCSIL-GFP载体以使其线性化。连接反应体系见表2。于16℃连接过夜。

表2 连接反应体系
1.2.4 转化 用氯化钙制备新鲜的大肠杆菌感受态细胞,从每种感受态细胞悬液中各取200 μ l转移到无菌的微量离心管中,每管加2 μ l连接液,混匀,在冰中放置30 min。将管放到42℃水浴中90 s,再快速将管转移到冰浴中,使细胞冷却1-2 min。每管加800 μ l SOC培养基。用水浴将培养基加温至37℃,然后将管转移到37℃摇床上,温育 45 min使细菌复苏。将150 μ l已转化的感受态细胞转移到含20 mmol/L MgSO4和 Amp 抗性(100 μ g/ml)的 LB 琼脂培养基上。将平板置于室温直至液体被吸收。倒置平皿,于37℃培养,16 h。
1.2.5 阳性克隆的鉴定
(1)阳性克隆的PCR鉴定PCR引物序列:Primer(+):5′-CCTATTTCCCATGATTCCTTCATA-3′,Primer:5′-GTAATACGGTTATCCACGCG-3′;PCR 反应体系 :10×buffer 2 μ l,dNTPs(2.5 mM)0.8 μ l,Primer(+)0.4 μ l,Primer(-)0.4 μ l,Taq polymerase 0.2 μ l,Template 1 μ l,ddH2O 补足 20 μ l;PCR 反应条件 :94℃,30 sec;94℃、30 sec,55℃、30 sec,72℃、30 sec,30 个循环;72℃,6 min。菌落PCR模版:在连接转化产物长出菌克隆表面沾一下 ,溶于10 μ l LB,混匀取 1 μ l作为模板;PCR产物行琼脂糖凝胶电泳观察。
(2)挑选阳性克隆送基因测序鉴定。
1.2.6 慢病毒的包装及滴度测定
(1)慢病毒包装细胞转染:转染前24 h,用胰蛋白酶消化对数生长期的293T细胞,以含10%血清的培养基调整细胞密度为1.2×107细胞/20 ml,重新接种于15 cm细胞培养皿,37℃、5%CO2培养箱内培养。24 h待细胞密度达70%-80%时即可用于转染。转染前2 h将细胞培养基更换为无血清培养基。向一灭菌离心管中加入所制备的各DNA溶液(pGC-LV 载 体 20 μ g,pHelper 1.0 载 体 15 μ g,pHelper2.0 载体 10 μ g),与相应体积的 Opti-MEM 混合均匀,调整总体积为2.5 ml,在室温下温育5 min。将Lipofectamine 2000 试剂轻柔摇匀,取 100 μ l Lipofectamine 2000试剂在另一管中与2.4 ml Opti-MEM混合,在室温下温育5 min。把稀释后的DNA与稀释后的Lipofectamine 2000进行混合。混合后,在室温下温育20 min,以便形成 DNA与 Lipofectamine 2000稀释液的转染复合物。将DNA与Lipofectamine 2000混合液转移至293T细胞的培养液中,混匀,于37℃,5%CO2细胞培养箱中培养。培养8 h后倒去含有转染混和物的培养基,每瓶细胞加入20 ml的PBS液以洗涤残余的转染混和物,然后倒去。每瓶细胞中加入含10%血清的细胞培养基 25 ml,于37℃、5%CO2培养箱内继续培养48 h。
病毒的收获及浓缩:收集转染后48 h的293T细胞上清液。于4℃,4 000 g离心10 min,除去细胞碎片。过滤、浓缩。分装后保存在病毒管中,-80度长期保存。取其中一支进行病毒生物学滴度测定。
(2)慢病毒滴度测定逐孔稀释法测定滴度:测定前一天,为293T细胞铺板,96孔板,每个孔加4×104个细胞,体积为100 μ l。根据病毒的预期滴度,准备7-10个无菌的Ep管。在每个管中加入90 μ l的无血清培养基。取待测定的病毒原液10 μ l加入到第一个管中,混匀后,取10 μ l加入到第二个管中。继续相同的操作直到最后一管。选取所需的细胞孔,吸去90 μ l培养基 ,丢弃。加入90 μ l稀释好的病毒溶液。放入培养箱培养。24 h后,加入完全培养基100 μ l。4天后,观察荧光表达情况。荧光细胞数随稀释倍数的增加而而减少。滴度计算:根据GFP荧光表达情况,病毒的滴度等于带有荧光的细胞数除以病毒原液量。
2 结果
2.1 人α-ASON重组慢病毒载体的鉴定结果
2.1.1 阳性克隆PCR鉴定
重组慢病毒载体构建成功后,转化感受态细胞,挑选阳性克隆进行PCR鉴定。PCR鉴定琼脂糖凝胶电泳结果如图1所示。PCR条带大小:连接入vshRNA片段的阳性克隆PCR片段大小为:343 bp(从载体中切掉24 bp);没有连接入vshRNA片段的空载体克隆PCR片段大小为:306 bp。
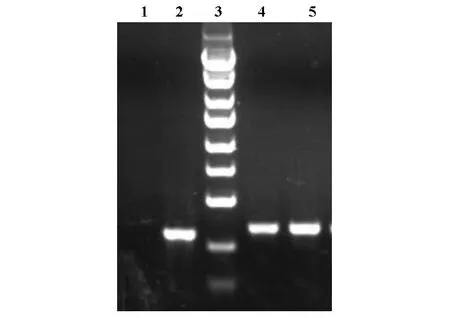
图1 阳性克隆PCR鉴定琼脂糖凝胶电泳图
2.1.2 阳性克隆测序结果分析 阳性克隆送基因测序结果如图2,与所需要的α-ASON序列完全一致。
2.2 人α-ASON重组慢病毒的滴度测定结果
在加入1×10-6μ l病毒原液的孔中观察到5个带有荧光的细胞,如图3。说明该孔中至少有5个病毒颗粒感染了细胞,则该病毒的滴度等于带有荧光的细胞数除以病毒原液量,就是5/(1×10-6)=5×106,单位为 TU/μ l,等于 5×109TU/ml。
3 讨论
β-地中海贫血的主要病理基础是由于α珠蛋白链的相对过剩,剩余的α珠蛋白链在红细胞内形成包涵体,导致红细胞膜的氧化损伤,造成红细胞破坏及骨髓的无效造血[4]。国外研究者认为适当下调内源性α-珠蛋白基因表达,减少α-珠蛋白肽链合成可能比增加β-珠蛋白肽链生成更有助于改善β-地中海贫血患者症状[5]。刘容容等[2,3]设计并筛选出能高效抑制α珠蛋白基因表达的α-ASON,并证实该α-ASON能有效地抑制体外培养的重型β-地中海贫血红系细胞α-珠蛋白基因表达,改善珠蛋白基因α/β+γ的比例失衡,使其红系细胞内过剩α-珠蛋白肽链的沉积明显减少。而α-ASON对于β-地中海贫血的体内试验尚未见报道。基因治疗载体系统是关系到基因治疗成败的重要因素之一。理想的载体应能有效的转移一个或多个功能基因,并且具有特异的靶向性,不被免疫系统识别,可以稳定、方便的扩增,高浓度的大量纯化,不会引起炎症,对受体和环境安全,并能按适当的调控模式表达携带的外源基因[6]。慢病毒为反转录病毒的一个亚科,它既可以感染分裂细胞又可感染非分裂细胞,研究表明慢病毒载体还具有转移的基因片段容量较大(9kb)、目的基因可整合至靶细胞基因组长期表达、免疫反应小等优点,且慢病毒载体对造血干细胞、免疫细胞等多种基因治疗的重要靶向细胞都有极好的细胞嗜性,因此慢病毒是基因治疗的较理想的载体;最为人们熟知的慢病毒是人免疫缺陷病毒(HIV),复制缺陷型HIV-1可被用于基因转移,HIV-1具有简单逆转录病毒的3个基因(gag、pol和env),此外还有6种辅助蛋白基因(tet、rev、vpr、nef和 vif)[6]。May等[7]在2000年首次报道用慢病毒载体对β-地中海贫血鼠模型进行基因治疗。Chen等[8]的研究显示,静止的造血干细胞被慢病毒载体有效转染,同时干细胞在体内自我更新和正常的种系特点不被损伤;用慢病毒载体有效的把基因导入到鼠干细胞,将使在诸如镰状细胞贫血和地中海贫血的血红蛋白疾病鼠模型,能进行基因治疗的直接试验。此外,绿色荧光蛋白(GFP)作为报告基因具有观察简单方便和直观的优点,只要有足够的表达,可以直接进行活观察,克服了其他报告基因需要底物及维持时间短的缺点[9,10]。

图2 阳性克隆基因测序结果

图3 人α-ASON重组慢病毒的滴度测定(加入 1×10-6μ l病毒原液)
实验结果显示,本实验成功构建了报告基因GFP和α-ASON序列融合的慢病毒载体三质粒系统,通过脂质体转染法将三质粒共转染293T细胞,包装产出病毒颗粒,并获得了5×109TU/ml较高的病毒滴度。PCR鉴定正确,基因测序结果与所需要的α-ASON序列完全一致。实验采用的慢病毒载体是以人类免疫缺陷型病毒(HIV)为基础发展起来的基因治疗载体,它可以在体内较长期的表达且安全性高,其为“自杀”性病毒,即病毒感染目的细胞后不会再感染其他细胞,也不会利用宿主细胞产生新的病毒颗粒。慢病毒中的毒性基因已经被剔除并被外源性目的基因所取代,属于假型病毒。实验采用的慢病毒载体系统由pGC-LV载体、pHelper 1.0载体和pHelper 2.0载体三质粒组成。pGC-LV载体中含有HIV的基本元件5′LTR和3′LTR以及其他辅助元件。通常根据不同的实验目的针对pGC-LV载体改造以进行启动子活性研究、基因表达研究、RNA干扰等研究。pHelper 1.0载体中含有HIV病毒的gag基因,编码病毒主要的结构蛋白;pol基因,编码病毒特异性的酶;rev基因,编码调节gag和pol基因表达的调节因子。pHelper 2.0载体中含有单纯疱疹病毒来源的VSV-G基因,提供病毒包装所需要的包膜蛋白。普通HIV-1载体借助其包膜表面的gpl20蛋白(由env基因编码)只能感染CIM(+)T细胞,当用水疱性口炎病毒(VSV)糖蛋白G或双嗜性小鼠白血病病毒(MLV)包膜蛋白取代HIV本身的包膜蛋白后,不仅进一步降低了HIV-1载体恢复成野生型病毒的可能同时由于VSV的包膜赋予HIV载体颗粒高度的稳定性,使其能够通过超速离心浓缩而达到高滴度;更重要的一点是包膜替换扩大了HIV-1载体的嗜性范围,使其几乎能感染所有组织来源的细胞如神经元细胞、肝细胞、肌纤维细胞、视网膜细胞等[11-14]。
目前大多数研究者都使用瞬时转染293T细胞的方式生产病毒载体。这种方法的缺点在于生产获得的病毒载体可能会出现有缺陷的基因组。为了生产高质量的载体,必须筛选合适的包装细胞系,而建立包装细胞系的最大难点在于某些病毒蛋白具有细胞毒性。目前,为了建立合适的慢病毒载体包装细胞系,许多科学家都在尝试使用诱导型或可调节的启动子来克服病毒蛋白的细胞毒作用[15,16]。本实验采用脂质体转染法将三质粒共转染293T细胞,包装病毒颗粒,效果较满意。而慢病毒载体滴度可以参考腺病毒载体滴度的检测方法[6],将病毒与293细胞共培养后,通过荧光显微镜观察GPF荧光表达或流式细胞仪检测GFP蛋白表达水平来分析病毒滴度。另外,还可以通过TaqMan PCR检测细胞中病毒拷贝数的方法检测滴度。由于荧光显微镜观察GPF荧光表达具有方便、简单和直观的优点,本研究采用此方法来分析计算病毒滴度。经测定,本研究人α-ASON重组慢病毒的滴度达到了5×109TU/ml,且基因测序结果与所需要的α-ASON序列完全一致。因此,本研究成功构建了介导人α-ASON的慢病毒载体并包装纯化慢病毒颗粒,为α-ASON对β地中海贫血基因治疗的体内试验奠定了基础。
[1]龙桂芳,张俊武主编.血红蛋白与血红蛋白病[M].广西科学技术出版社,2003.
[4]Shinar E,Rachmilewitz EA.Oxidative denaturation of red blood cells in thalassemia[J].Semin Hematol,1990,27(1):70.
[5]Thein SL.Pathophysiology of{beta}thalassemia-A guide to molecular therapies.Hematology Am Soc Hematol Educ Program,2005:31-7.
[6]陈金中,薛京伦主编.载体学与基因操作[M].北京:科学出版社,2007.
[7]May C,Rivella S,Callegari J,et al.Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin[J].Nature,2000,406:82.
[8]Chen W,Wu X,Levasseur DN,et al.Lentiviral vector transduction of hematopoietic stem cells that mediate long-term reconstitution of lethally irradiated mice[J].Stem Cells,2000,18(5):352.
[9]Tomioka R,Rockland KS.ImprovedGolgi-like visualization in retrogradely projecting neurons after EGFP-adenovirus infection in adult rat and monkey[J].J Histochem Cytochem,2006,54(5):539.
[10]Dijon M,Torne-Celer C,Moreau T,et al.Expression and recombination of the EGFP and EYFP genes in lentiviral vectors carrying two heterologous promoters[J].Cytotherapy,2005,7(5):417.
[11]Naldini L,Blomer U,Gage FH,et al.Efficient transfer,integration,and sustained long-ter m expression of the transgene in adult rat brains injectedwith a lentiviral vector[J].Proc Natl Acad Sci USA,1996,92(21):11382.
[12]Zuferey R,Nagy D,Mandel RJ,et al.Mutiply attenuated lentiviral vector achieved eficient gene delivery in vivo[J].Nat Biotechnol,1997,15(9):871.
[13]Miyoshi H,Smith KA,Mosier DE,et al.Transduction of human CD34+cells that mediate long-term engraftment of NOD/SCID mice by HIV vectors[J].Science,1999,29:283(5402):682.
[14]Kafri T,Blomer U,Peterson DA,et al.Sustained expression of genes delivered directly into liver andmuscle by lentiviral vectors[J].Nat Genet,1997,17(3):314.
[15]Kaul M,Yu H,Ron Y,et al.Regulated lentiviral packaging cell line devoid of most viral cis-acting sequences[J].Virology,1998,249(1):167.
[16]Kafri T,van Praag H,Ouyang L,et al.A packaging cell line for lentivirus vectors[J].J Virol,1999,73(1):576.
Construct recombinant lentiviral vectors carrying human α-antisense oligonucleotide
LAI Ying-hui,LAI Yong-rong,YANG Gao-hui.(Department of Hematology,the First Affiliated Hospital,Guangxi Medical University,Nanning530021,China)
ObjectiveTo construct recombinant lentiviral vectors carrying human α-antisense oligonucleotide.MethodsAccording to the human α-antisense oligonucleotide sequence,pGCSIL-vshRNA-GFP plasmid was constructed by double restriction enzyme digestion and ligation,and then the plasmidwas transformed into E.coliDH5α.Purified pGCSIL-vshRNA-GFP plasmids from the positive clones was confirmed by PCR and sequencing.293T cells were cotransfected with lentiviral vector pGCSIL-vshR NA-GFP,pHelper 1.0 and pHelper 2.0 by Lipofectamine 2000 to produce lentivirus.The titer of virus was tested according to the expression level of green fluorescent protein.ResultsThe exogenous gene sequence of the recombinant plasmids was completely in accordance with that of the human α-antisense oligonucleotide.The titer of concentrated virus was 5×109TU/ml.ConclusionThe recombinant lentiviral vectors carrying humanα-antisense oligonucleotide are successfully constructed.
Lentiviral vector;Antisense oligonucleotide;Gene therapy
R392
A
1007-4287(2010)09-1358-05
国家自然科学基金资助项目(30860307),广西研究生教育创新计划资助项目
*通讯作者
2010-05-10)
猜你喜欢
化工设计通讯(2022年6期)2023-01-02
检验医学与临床(2022年18期)2022-09-27
中国卫生标准管理(2022年14期)2022-09-13
北京生物医学工程(2022年4期)2022-08-18
转化医学杂志(2022年1期)2022-03-07
生物工程学报(2018年5期)2018-06-11
华人时刊(2017年21期)2018-01-31
中国比较医学杂志(2018年5期)2018-01-22
中国畜禽种业(2016年2期)2016-01-27
中国生化药物杂志(2015年7期)2015-07-07
