
寡核苷酸药物及寡核苷酸制备技术研究进展
2018-06-11 02:07:30汪小龙咸静女陈刚彭海波
生物工程学报 2018年5期
汪小龙,咸静女,陈刚,彭海波
中国海洋大学 海洋生命科学学院 生物工程系,山东 青岛 266003
对于癌症、病毒性感染等疾病,迄今临床上仍缺少理想的特效药物。随着人类及重要模式生物基因组测序的完成,以及功能基因组学及蛋白质组学研究的深入,与疾病相关的分子靶标不断被发现和认识,为基因治疗提供了前提。在过去的几十年里,人工合成的寡核苷酸已经广泛应用于靶向基因治疗的研究,主要包括反义寡核苷酸(Antisense oligonucleotides, ASODN)、小干扰 RNA(Small interference RNA, siRNA)、转录因子诱饵(Decoy)、核酶 (Ribozyme)、脱氧核酶 (DNAzyme)、反基因 (Antigene)、CpG寡核苷酸和核酸适配体(Aptamer) 等。其中ASODN和siRNA是最常用的基因调控工具,获得广泛应用,并已被开发为基因治疗药物。目前,寡核苷酸主要采用化学方法合成,其初产品纯度低,难以纯化,大量制备成本十分高昂,大大限制了寡核苷酸药物的研究和应用。本文综述了寡核苷酸药物的类别、作用原理及其特点,指出了寡核苷酸常规化学制备方法存在的问题,并介绍了新型核酸扩增及寡核苷酸制备方法的研究进展。
1 寡核苷酸药物的类别及其作用原理
1.1 反义寡核苷酸
反义寡核苷酸 (Antisense oligonucleotides,ASODNs) 是指人工合成的、与特定基因互补的寡核苷酸 (DNA或 RNA) 及其类似物,长度为13−25个碱基的核酸片段,它可以通过碱基互补配对原则结合于靶基因或靶RNA上,从而抑制基因的表达[1]。很多遗传性、代谢性和病毒性疾病都是由于基因表达异常引起的。反义寡核苷酸作为药品的原理是利用碱基互补配对原则与特定的信使RNA (Messenger RNA,mRNA) 进行配对,抑制其表达,阻断遗传信息从 DNA传递到蛋白质。根据核酸杂交原理,反义寡核苷酸能与特定的基因、病毒核酸或其转录产物结合,特异性抑制致病基因或病毒基因的表达。通过反义寡核苷酸抑制致癌基因或病毒的关键编码基因,可特异性抑制肿瘤细胞增殖生长并诱导细胞凋亡[2]。靶向mRNA或 microRNA的反义寡核苷酸,已被广泛用作分子生物学的研究工具,以特异性和选择性地下调特定基因的表达。基因表达的产物是蛋白质,传统小分子药物主要作用于致病蛋白质,而反义寡核苷酸药物则直接作用于致病基因本身,因此比小分子化学药物更具选择性,而且具有高效、低毒、稳定有效、不易产生耐药性等优点,是理想的抗癌和抗病毒药物[3]。第一个通过美国食品及药品管理局 (Food and Drug Administration,FDA) 咨询委员会认证并应用于临床的反义核酸药物是用于治疗艾滋 (AIDS) 病人巨细胞病毒感染的视网膜炎的福米韦生 (Fomivirsen或Virtravene)[4]。然而,早先的实验室或临床研究中一些反义核酸稳定性、特异性和有效性不高,并且存在严重的毒副作用,因此学术界一度对反义核酸药物产生质疑,并导致上世纪末反义核酸药物的研发相对较为缓慢。
然而,近年来一些高效的新型反义核酸药物的出现,使反义核酸药物研究得以复兴。反义寡核苷酸作为用于抗肿瘤、抗病毒治疗的药物,是目前最有应用前景的基因靶向治疗药物。目前有数十种反义寡核苷酸药物正在进行临床试验或动物实验,针对包括癌症、肥胖、心血管疾病、精神病、代谢性疾病和病毒感染等。靶向作用于miRNA或 mRNA的反义寡核苷酸预计能用于治疗多种人类疾病。2013年 1月美国 ISIS公司和Genzyme公司合作的一大成果——反义寡核苷酸药物Mipomersen (商品名KYNAMRO™) 通过美国FDA认证[5-6]。Mipomersen是合成的烷基修饰反义寡核苷酸辅助降脂药物,通过抑制载脂蛋白ApoB-100的mRNA,以降低家族性高胆固醇血症(HoFH) 患者的低密度脂蛋白胆固醇 (LDL-C)、载脂蛋白B (Apo B)、总胆固醇 (TC) 和非高密度脂蛋白胆固醇 (非高密度脂蛋白C)[7-9]。
如图1所示,针对成熟mRNA,既可以设计反义寡核苷酸,使其与mRNA结合形成杂合体,利用RNase H识别并酶解DNA:RNA杂合体的作用,使mRNA降解,从而抑制基因表达;也可以针对mRNA 5′-非翻译区设计反义寡核苷酸,使其翻译受阻。另外,针对前体mRNA,还可以在内含子与外显子剪切位点处设计反义寡核苷酸,使其不能正常剪切,称为剪切捕获 (Splicing arrest),此方法已经成功应用于杜氏肌营养不良 (Duchenne muscular dystrophy,DMD) 的基因治疗[10-14]。DMD 是抗肌萎缩蛋白 (肌营养蛋白) 外显子跳读 (Exon skipping) 引起的。2016年9月19日,FDA批准Sarepta Therapeutics公司的反义RNA药物Eteplirsen (Exondys 51),用于治疗外显子51跳读型杜氏肌营养不良。Eteplirsen通过静脉注射给药,可帮助51号外显子跳读DMD患者合成一些抗肌萎缩蛋白,延缓疾病进程,是FDA批准的首个DMD药物。
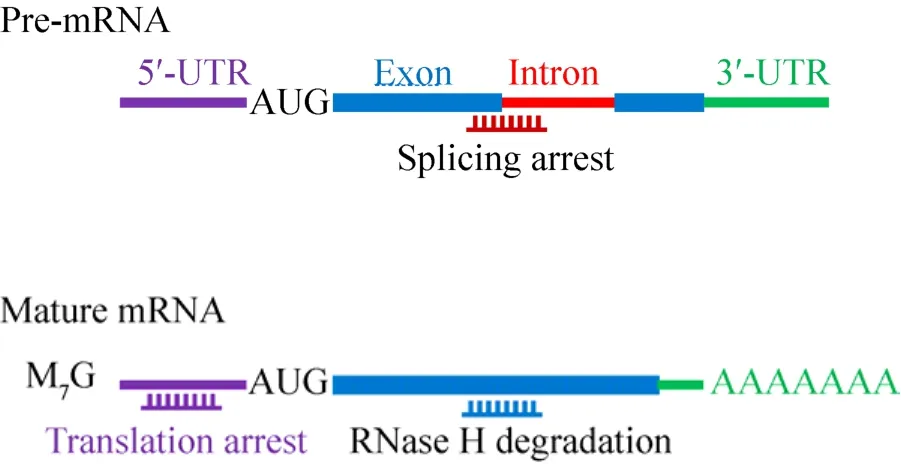
图1 反义寡核苷酸设计原理示意图Fig. 1 Schematic diagram of antisense oligonucleotide.
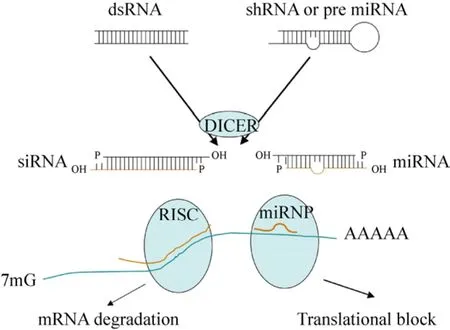
图2 RNA干扰原理示意图 (图片引自文献[15])Fig. 2 Schematic diagram of RNA interference[15].
1.2 小分子干扰RNA
小分子干扰RNA (Small interfering RNA, siRNA)是一类长度在20−25 bp的双链RNA片段。siRNA通过与特定的核酸序列互补而影响特定基因的表达,在生物体内许多代谢过程中扮演重要角色。RNA干扰的原理如图2所示,siRNA通过结合到靶标 mRNA上使其降解或抑制其翻译[15]。Hamilton等于1999年首先在Science杂志报道发现了 siRNAs在植物转录后基因沉默中的作用[16]。Lagos-Quintana等2003年在Nature杂志报道了化学合成的 siRNA可以在哺乳动物细胞中引起RNAi作用[17]。随后一系列的发现激发了学术界和产业界应用 RNAi技术在生物医学研究和药物开发上的热情。作为基因治疗药物研究的热点,小分子干扰RNA研究方兴未艾,然而至今尚未有siRNA药物开发成功上市。究其原因,是由于siRNA与靶标的结合并非一一对应,而是具有一定的非特异性,容易产生脱靶效应[18-19]。与反义寡核苷酸相比,siRNA既有级联放大效应带来的作用显著、需要剂量低的优点,但又有稳定性差、容易被降解和难以被输送到靶细胞的缺点。
1.3 核酸适配体
核酸适配体 (Aptamer) 是一类由化学合成的单链RNA或单链DNA寡核苷酸组成的小分子核酸配基[20]。其原理如图3所示,核酸适配体能够折叠形成高级结构,并通过识别特异的三维结构,与其靶标进行相互作用,具有高特异性和亲和性,故而又有化学抗体之称。
与普通的蛋白抗体相比,核酸适配体有诸多优点:一是由于其分子量较低 (8–25 kDa),因此能够快速渗透组织;二是适配体在体内几乎不会引起免疫反应[21];三是由于核酸适配体分子本质上具有很强的热稳定性;四是核酸适配体识别范围非常广,能够识别离子、药物、毒素、蛋白质[22]、病毒[23]、细菌[24]、癌细胞[25],甚至肿瘤和组织[26-27]。
近年来,核酸适配体已被应用于临床研究,例如能与血管内皮生长因子特异性结合的一种RNA适配体药物Macugen已被美国FDA批准用于老年湿性黄斑变性 (Wet age-related macular degeneration) 以及家族性渗出性玻璃体视网膜病变 (Familial exudative vitreoretinopathy) 的治疗[28-30]。另外,如表1所示,目前还有多种适配体药物处于临床试验阶段。

表1 处于临床试验阶段的治疗性核酸适配体Table 1 Therapeutic aptamers in clinical trials stage
1.4 三股螺旋寡核苷酸
通过特异性识别 DNA双螺旋中的多聚嘌呤或多聚嘧啶区域,寡核苷酸能够与DNA双螺旋的大沟或小沟结合形成局部三股螺旋结构[31-33],被称为三股螺旋寡核苷酸 (Triplex-forming oligonucleotide,TFO)。如图4所示,TFO能够对目标基因有效定位及封锁,在转录水平抑制基因表达[34],或在人体细胞中直接对基因组 DNA进行修饰以修复遗传损伤[35],有着重要应用潜力和研究价值。
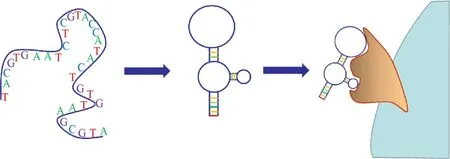
图3 核酸适配体识别靶标示意图Fig. 3 Schematic diagram of aptamer binding to its target.
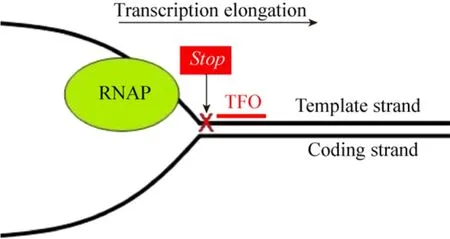
图 4 三股螺旋寡核苷酸抑制转录延伸 (图片引自文献[34])Fig. 4 Triplex-forming oligonucleotide block the transcription elongation[34].
1.5 转录因子诱饵寡核苷酸
转录因子诱饵寡核苷酸 (Decoy ODN) 是新一代强有力的反义基因策略,用于治疗多种疾病,并可确定某种特定基因表达的分子机制。如图 5所示,诱饵寡核苷酸是双链的,其序列与顺式作用元件的序列是一致的,因此能够通过竞争性结合抑制转录因子的功能作用。因此,用于转染的双链decoy ODN能够减弱基因组中真正的顺式作用元件和反式作用元件的相互作用,导致结合在内生性顺式元件上的反式因子被移除,控制目标基因的表达,从而发挥基因治疗作用[36]。
转录诱饵寡核苷酸已被试用于多种人类肿瘤和遗传性疾病的基因治疗。Morishita研究组以转录因子E2F的转录诱饵来抑制动脉粥样硬化和动脉内膜增生相关基因的表达,以治疗冠心病和血管增殖性心血管疾病[36-38]。如表2所示,目前还有很多转录诱饵寡核苷酸正在进行临床研究,预期具有良好的应用前景。
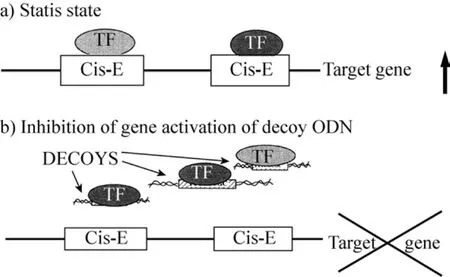
图 5 诱饵寡核苷酸法抑制转录因子 (TF) 的功能(图片引自文献[36])Fig. 5 The decoy oligonucleotide approach to block the function of the transcription factor (TF)[36].
1.6 核酶和脱氧核酶
早在1981年,Zaug等发现四膜虫rRNA的前体在没有蛋白质酶的催化作用下能专一性地催化寡核苷酸底物的连接与切割,证明RNA分子也具有催化活性,称之为核酶 (Ribozyme)[39]。直到20世纪80年代末90年代初,核酶的结构才有了初步的了解,是一种简单的催化结构域——锤头结构 (Hammerhead ribozyme)[40]。核酶可以很容易地用化学方法合成,随着对核酶功能以及结构研究的不断深入,核酶在抑制目的基因表达的研究过程中得到了广泛的应用。
由于核酶具有不稳定性,且利用化学修饰的过程比较复杂,1997年Santoro等通过体外筛选的方法设计出了一种依赖Mg2+离子、可特异性切割RNA的脱氧核酶 (DNAzyme)[41]。如图6所示,DNAzyme有两条任意可变的结合臂 (armⅠ和armⅡ),中间的催化中心含有 14–15个碱基的保守序列。其中10–23 DNAzyme对底物RNA的要求是切割部位核心序列含有RY (R=A/G, Y=C/U)。与核酶相比,DNAzyme更易合成,对化学物质或核酸酶有较高的抵抗力,而且 DNAzyme对于DNA:RNA 杂合链与RNA:RNA 双链在酶的评价指标方面 (如底物的选择性、靶向性、酶的转换率、催化效率等),与核酶相比均有所提高[41]。DNAzyme目前已经广泛用于治疗肝炎[42]、艾滋病[43]、癌症[44-46]等重大人类疾病的研究中。
2 寡核苷酸大规模制备技术存在的问题
规模化生产是药物开发的基础,这一点对于反义寡核苷酸药物尤为重要。反义寡核苷酸药物是目前最为成熟的寡核苷酸药物,但由于其没有级联放大作用,在临床和动物实验中使用剂量大,高达 25−50 mg/kg[47]或 800−1 000 mg/周[48]。因此,作为药物的寡核苷酸必须能够规模化生产,合成克级甚至千克级的寡核苷酸,并要降低成本,才有可能开展临床和动物实验。目前寡核苷酸几乎都是使用化学方法合成,即采用固相亚磷酰胺三酯法合成,并用高效液相色谱分离纯化。常规的DNA合成仪只能合成毫克级的寡核苷酸,大规模DNA合成必须采用克级和千克级DNA合成仪,如GE Healthcare公司生产的ÄKTA OligoPilot系列。目前仅有少数大型国际生物制药公司,如美国ISIS公司和丹麦的Santaris公司,才有能力合成千克级的寡核苷酸,开展反义药物的大规模临床研究。国内仅有军事医学科学院王升启等开展了寡核苷酸大规模合成及动物实验[49],但目前尚无核酸药物上市或进入临床实验。究其主要原因,大规模合成寡核苷酸,仪器设备成本过于高昂,一直是寡核苷酸药物研究的瓶颈。
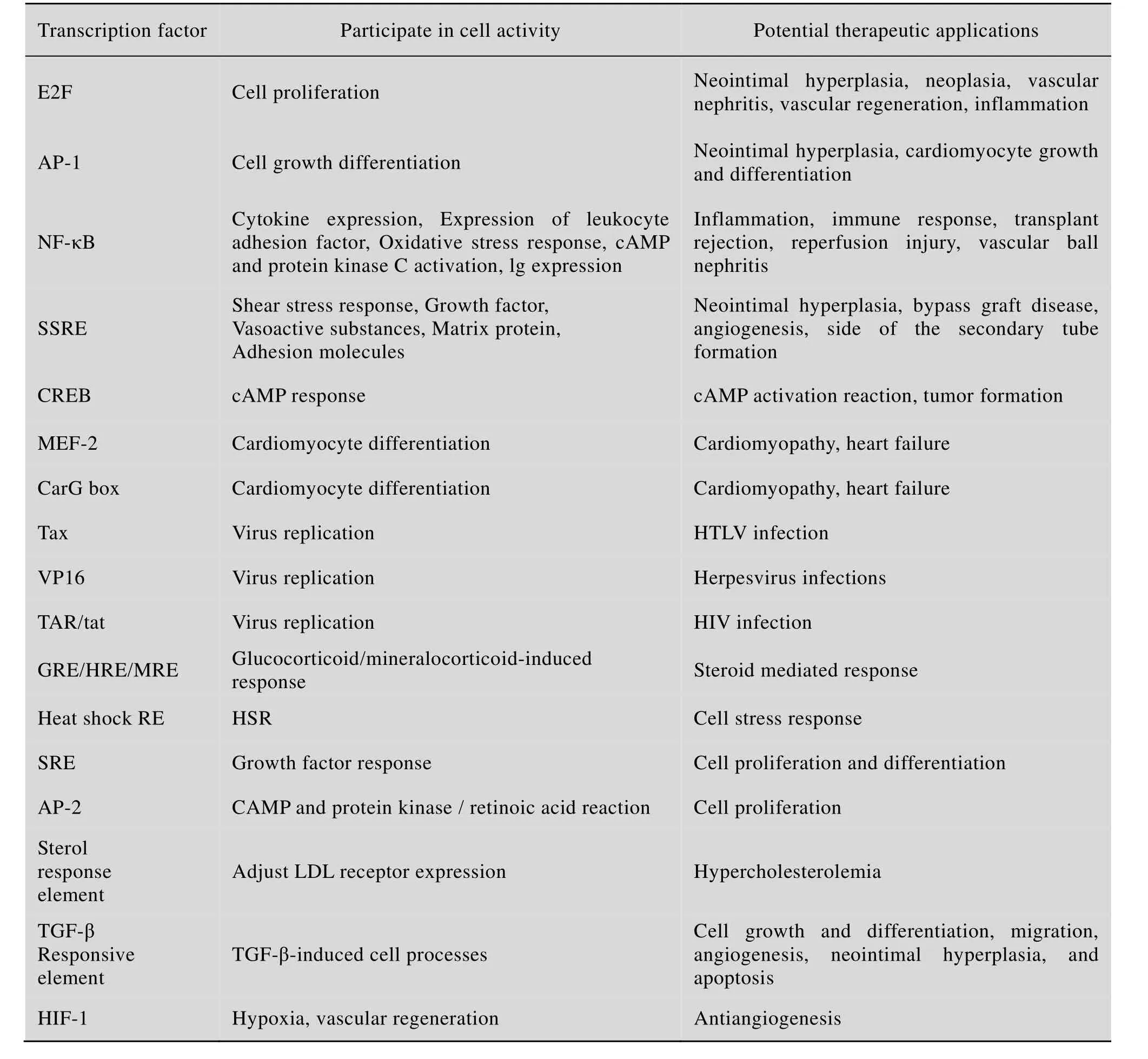
表2 用诱饵寡核苷酸作为基因治疗的潜在靶标转录因子Table 2 Transcription factors as potential therapeutic targets for gene manipulation by using decoy oligonucleotides
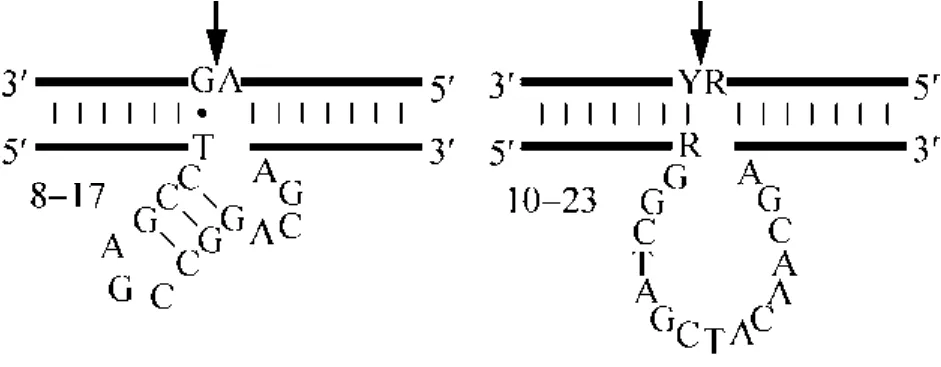
图 6 两种 DNAzyme (8–17和10–23) 与底物RNA结合和切割位点示意图 (图片引自文献[41])Fig. 6 The binding and cleaving of the RNA substrates by two different (8–17 and 10–23) DNAzymes[41].
另外,化学合成寡核苷酸初产品被杂质和截短的错误序列 (又叫失败序列) 所污染,使产物纯化十分困难。产生这些失败序列的原因是在合成过程中脱三苯甲基 (Detritylation)、耦合 (Coupling)、硫化 (Sulfurization) 或保护 (Capping) 等反应不完全造成的,因此,必须对寡核苷酸粗产品进行纯化。然而合成寡核苷酸中最难以除去的杂质是大量 (n–1) 缺失序列 (与完整长度为n的寡核苷酸相比仅相差一个核苷酸)。对于长度为18–21碱基对的寡核苷酸,用高效液相色谱法几乎无法从产品中完全除去 (n–1) 缺失序列[50]。此外,合成过程需要采用多种有害化学试剂,例如去保护试剂——三氯乙酸 (TCA) 或二氯乙酸 (DCA),必须溶解在卤化溶剂中 (通常是二氯甲烷)。二氯甲烷具有高毒性、高挥发性和致癌性。因此,大规模的寡核苷酸生产不仅成本高昂,而且涉及到使用和处理危险化学品,对环境和人体存在潜在的威胁。
3 核酸扩增技术应用于制备寡核苷酸
寡核苷酸在现代分子生物技术和生物制药研究中应用十分广泛。对具有特定序列的目标寡核苷酸进行扩增,不仅是一个需要克服的技术难题,而且在生物医药科学研究中具有重要的应用价值。商业化药物生产需要大量 (以千克计) 的寡核苷酸。传统的化学合成寡核苷酸不仅成本高昂,而且初产品纯度低、难以纯化,十分不利于临床研究和大规模药物生产。因此,开发一种简单、经济和安全的寡核苷酸大规模工业化生产方法已成为必要,而采用生物技术,用核酸扩增方法制备寡核苷酸无疑是最佳方案。
聚合酶链式反应 (Polymerase chain reaction,PCR) 技术自诞生以来[51],一直是核酸扩增的主要方法。PCR和逆转录PCR技术在用于扩增有足够长度的DNA和RNA时简单而有效。但无法用PCR技术直接扩增短链寡核苷酸和siRNA。因为它们往往只有18–30个核苷酸的长度,无法设计一对特异性PCR引物。用PCR来扩增寡核苷酸或 siRNA,首先必须设法将目标延长 (如加接头或使用通用引物),如此不可避免地会在产物中引入非目标序列。这在检测分析方法中没有问题,但对于制备方法而言,非常不利于后期分离纯化。而且由于底物是修饰性dNTPs,价格非常昂贵,产物中如果含有非目标序列,会造成底物 (dNTPs)的浪费,是难以接受的。另外,更重要的是PCR只是将寡核苷酸引物延伸,PCR扩增产物的分子数不可避免地受输入寡核苷酸引物分子数的限制,因此PCR只能实现DNA分子量增加,而无法使寡核苷酸分子数目增加。因此,PCR技术不适合进行短链寡核苷酸或siRNA的大量制备。
近年来,基于核酸扩增技术的检测和诊断方法已获得广泛应用,给临床诊断提供了快速、灵敏和准确的方法。因此,国内外众多学者不断致力于改进和探索新型核酸扩增技术,已经建立了多种多样的新型核酸扩增方法。按照扩增过程中温度是否变化来划分,核酸扩增方法可分为热循环扩增和恒温扩增两大类。热循环扩增主要包括经典的核酸扩增技术——PCR[51-52]和连接酶链式反应 (Ligase chain reaction, LCR)[53-55],而恒温扩增主要包括链置换扩增 (Strand displacement amplification, SDA)[56-58]、滚环扩增 (Rolling circle amplification, RCA)[59-61]、环介导扩增 (Loop mediated amplification, LAMP)[62-63]、依赖解旋酶扩增(Helicase-dependent amplification, HDA)[64-65]、依赖核酸序列的扩增 (Nucleic acid sequence based amplification,NASBA)[66-67],等等。
这些DNA扩增技术和PCR一样,都需设计并采用至少一对化学合成的寡核苷酸作为引物,不可避免地会在产物中引入非目标序列。此外,这些方法只是将寡核苷酸引物延伸,其扩增产物的分子数也同样不可避免地受输入寡核苷酸引物浓度的限制,二次扩增必须加入新的寡核苷酸引物。因而一般也都不适合大量制备长度约20个碱基左右的寡核苷酸。
目前,已报道的专门用于扩增和制备短寡核苷酸的方法极少。指数扩增反应 (EXPAR) 是一个极为快速的恒温反应[68],能够在10 min内快速扩增和制备目标寡核苷酸。EXPAR依赖于切刻内切酶Nt.BstNBI和具有链置换活性的DNA聚合酶,因此又被命名为切刻酶扩增反应 (Nicking enzyme amplification reaction, NEAR)[69]。然而,目前EXPAR的应用十分有限,因为据报道该反应存在严重的非特异性扩增[70]。此外,该反应产物是单链的,仅对目标链进行扩增,而其模板 (反义链)并未得到扩增。并且,EXPAR反应无法进行二次扩增,产量还是会受输入模板分子数目的限制,因此也不适合大量制备寡核苷酸。另外,滚环复制 (Rolling circle replication, RCR) 方法已被用于扩增环形DNA,可用于制备寡核苷酸[71]。然而,每轮 RCR扩增之前都必须要进行费时费力的人工酶切、连接和重新环化,使之不适合进行自动化的大规模寡核苷酸制备。最近,Ducani等在Nature Method杂志报道改进版的RCR方法,称为 Monoclonal stoichiometric (MOSIC) 方法[72]。MOSIC方法依赖于具有链置换活性的Phi29 DNA聚合酶,以及特殊的内切酶 BseGI,可按照预设的比例产生多个目标序列的单链寡核苷酸,预期可用于 DNA纳米材料的制备。但目前该方法尚未实现制备带有修饰基团的药用寡核苷酸。
4 新型核酸扩增方法——PEAR技术及其在寡核苷酸制备中的应用
如前所述,现有核酸扩增技术在应用于寡核苷酸扩增时均存在一定的困难。2010年Wang等研究开发了一种新型热循环酶促生物化学反应,称为聚合酶-内切酶扩增反应 (PEAR)[73],用于扩增和大量制备寡核苷酸。PEAR技术采用含有重复目标序列及酶切位点的双链DNA作为“种子”,通过独特的“滑动-切割机制”,用耐热 DNA聚合酶延伸,耐热限制性内切酶切割,在热循环控制下,使寡核苷酸等小分子核酸能够像病毒一样自我复制,扩增特定的目标寡核苷酸。PEAR扩增产物的分子数能够以指数模式持续增加,并且不受输入初始“种子”寡核苷酸分子数的限制。PEAR产物无需经过任何处理,无需加入新的引物和模板,直接作为“种子”进行二次扩增。
PEAR反应DNA聚合酶和内切酶的选择范围很大,可制备各种天然和修饰寡核苷酸。目前用PEAR技术已经成功扩增和制备了带有硫代[74]和氟代[75]修饰基团的寡核苷酸。经高效液相色谱和质谱联用技术检测,表明PEAR产物中目标寡核苷酸所占比例很高。由于避免了产生 (n–1) 缺失序列,纯化过程十分容易。对于双链寡核苷酸而言,则无需进行高效液相色谱分离,用普通柱层析纯化后即可。因而,PEAR技术特别适合制备双链寡核苷酸,如转录因子诱饵。
PEAR技术具有操作简便、高效和稳定的特点,能够快速、准确、稳定地扩增寡核苷酸,产物纯度高。PEAR所采用的DNA聚合酶和内切酶等主要原材料都是纯生物制剂,生产过程安全无污染。并且,用PEAR方法制备寡核苷酸不需要昂贵的大规模 DNA合成仪,而是采用价格低廉的热循环仪。因此,大规模生产的设备和材料费用均可大大降低。PEAR作为一种简单而高效的寡核苷酸扩增和制备方法,能够解决上述寡核苷酸生产中所存在的诸多问题。
5 结论
寡核苷酸药物研究方兴未艾,应用前景广阔。寡核苷酸通常采用化学方法合成,但化学合成寡核苷酸会出现错误,纯度较低,而纯化十分困难。而且,由于天然的寡核苷酸在体内会被很快降解,具有一些毒副作用,因此作为药物的寡核苷酸必须带有修饰基团,增强寡核苷酸在体内的稳定性。大规模化学合成寡核苷酸的成本十分高昂,大大限制了寡核苷酸药物的研究和应用。目前已经出现了多种核酸扩增方法,但均不适用于大量制备寡核苷酸。新的寡核苷酸扩增方法“聚合酶-内切酶扩增反应” (PEAR) 使寡核苷酸等小分子核酸能够在两种耐热酶的催化作用下,通过热循环进行自我复制,并实现指数扩增。这种新的寡核苷酸生产方法具有很多优点,能解决目前寡核苷酸制造中的诸多问题。该方法采用大容量热循环仪生产寡核苷酸,设备成本低,适合大量生产修饰寡核苷酸。PEAR技术是拥有自主知识产权的核酸扩增技术,很有可能用于大量生产成分更纯、成本低廉的寡核苷酸类药物,有助于推动寡核苷酸药物的研究和应用。
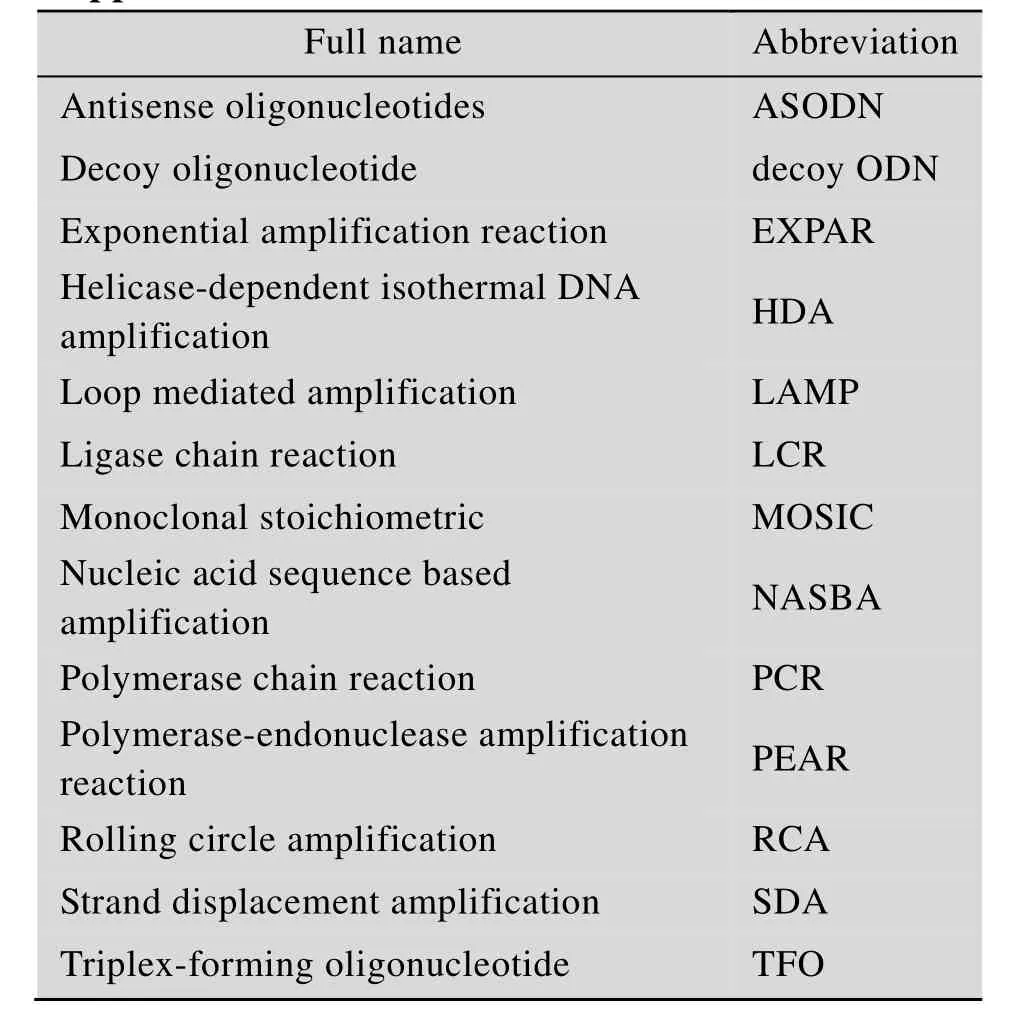
附表1 各名词全称及缩写归纳表Appendix 1 Abbreviations
[1]DeVos SL, Miller TM. Antisense oligonucleotides:treating neurodegeneration at the level of RNA.Neurotherapeutics, 2013, 10(3): 486–497.
[2]Berg RW, Ferguson PJ, Vincent MD, et al. A"combination oligonucleotide" antisense strategy to downregulate thymidylate synthase and decrease tumor cell growth and drug resistance. Cancer Gene Ther, 2003,10(4): 278–286.
[3]Selvam MP, Buck SM, Blay RA, et al. Inhibition of HIV replication by immunoliposomal antisense oligonucleotide. Antiviral Res, 1996, 33(1): 11–20.
[4]Listed N. Fomivirsen approved for CMV retinitis: first antisense drug. Aids Treat News, 1998, (302): 7.
[5]Haddley K. Mipomersen sodium: a new option for the treatment of familial hypercholesterolemia. Drugs Today(Barc), 2011, 47(12): 891–901.
[6]Hair P, Cameron F, Mckeage K. Mipomersen Sodium:First Global Approval. Drugs, 2013, 73(5): 487–493.
[7]McGowan MP, Tardif JC, Ceska R, et al. Randomized,placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PLos ONE, 2012, 7(11):e49006.
[8]Santos RD, Duell PB, East C, et al. Long-term efficacy and safety of mipomersen in patients with familial hypercholesterolaemia: 2-year interim results of an open-label extension. Eur Heart J, 2015, 36(9): 566–575.
[9]Duell PB, Santos RD, Kirwan BA, et al. Long-term mipomersen treatment is associated with a reduction in cardiovascular events in patients with familial hypercholesterolemia. J Clin Lipidol, 2016, 10(4):1011–1021.
[10]Echigoya Y, Lim KRQ, Trieu N, et al. Quantitative antisense screening and optimization for exon 51 skipping in Duchenne muscular dystrophy. Mol Ther,2017, 25(11): 2561–2572.
[11]Jirka SMG, Winter TD, Meulen BVD, et al. Evaluation of 2′-Deoxy-2′-fluoro antisense oligonucleotides for exon skipping in Duchenne muscular dystrophy. Mol Ther Nucleic Acids, 2015, 4(12): e265.
[12]Kendall GC, Mokhonova EI, Moran M, et al. Dantrolene enhances antisense-mediated exon skipping in human and mouse models of Duchenne muscular dystrophy. Sci Transl Med, 2012, 4(164): 164ra160.
[13]Aartsma-Rus A, Janson AA, van Ommen GJ, et al.Antisense-induced exon skipping for duplications in Duchenne muscular dystrophy. BMC Med Genet, 2007,8: 43.
[14]Incitti T, de Angelis FG, Cazzella V, et al. Exon skipping and duchenne muscular dystrophy therapy: selection of the most active U1 snRNA antisense able to induce dystrophin exon 51 skipping. Mol Ther, 2010, 18(9):1675–1682.
[15]Sledz CA, Williams BR. RNA interference in biology and disease. Blood, 2005, 106(3): 787–794.
[16]Hamilton AJ, Baulcombe DC. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science, 1999, 286(5441): 950–952.
[17]Lagos-Quintana M, Rauhut R, Meyer J, et al. New microRNAs from mouse and human. RNA, 2003, 9(2):175–179.
[18]Di Rocco G, Verdina A, Gatti V, et al. Apoptosis induced by a HIPK2 full-length-specific siRNA is due to off-target effects rather than prevalence of HIPK2-Deltae8 isoform. Oncotarget, 2016, 7(2):1675–1686.
[19]Fedorov Y, Anderson EM, Birmingham A, et al.Off-target effects by siRNA can induce toxic phenotype.RNA, 2006, 12(7): 1188–1196.
[20]Conrad RC, Giver L, Tian Y, et al.In vitroselection of nucleic acid aptamers that bind proteins. Methods Enzymol, 1996, 267: 336–367.
[21]Kong HY, Jonghoe B. Nucleic acid aptamers: new methods for selection, stabilization, and application in biomedical science. Biomol Ther (Seoul), 2013, 21(6):423–434.
[22]Dua P, Kim S, Lee DK. Nucleic acid aptamers targeting cell-surface proteins. Methods, 2011, 54(2): 215–225.
[23]Zhang Z, Zhang J, Pei X, et al. An aptamer targets HBV core protein and suppresses HBV replication in HepG2.2.15 cells. Int J Mol Med, 2014, 34(5):1423–1429.
[24]Meyer MM, Roth A, Chervin SM, et al. Confirmation of a second natural preQ1 aptamer class in Streptococcaceae bacteria. RNA, 2008, 14(4): 685–695.
[25]Kim YH, Sung HJ, Kim S, et al. An RNA aptamer that specifically binds pancreatic adenocarcinoma up-regulated factor inhibits migration and growth of pancreatic cancer cells. Cancer Lett, 2011, 313(1):76–83.
[26]Duan M, Long Y, Yang C, et al. Selection and characterization of DNA aptamer for metastatic prostate cancer recognition and tissue imaging. Oncotarget, 2016,7(24): 36436–36446.
[27]Li S, Xu H, Ding H, et al. Identification of an aptamer targeting hnRNP A1 by tissue slide-based SELEX. J Pathol, 2009, 218(3): 327–336.
[28]Pegaptanib sodium (Macugen) for macular degeneration.Med Lett Drugs Ther, 2005, 47(1212): 55–56.
[29]Tobin KA. Macugen treatment for wet age-related macular degeneration.Insight, 2006, 31(1): 11–14.
[30]Quiram PA, Drenser KA, Lai MM, et al. Treatment of vascularly active familial exudative vitreoretinopathy with pegaptanib sodium (Macugen). Retina, 2008, 28(3 Suppl): S8–12.
[31]Kim HG, Miller DM. Inhibition ofin vitrotranscription by a triplex-forming oligonucleotide targeted to human c-myc P2 promoter. Biochemistry, 1995, 34(25):8165–8171.
[32]Okada T, Yamaguchi K, Yamashita J. Triplex-forming oligonucleotide binding represses transcription of the human c-erbB gene in glioma. Growth Factors, 1994,11(4): 259–270.
[33]Praseuth D, Guieysse AL, Itkes AV. Unexpected effect of an anti-human immunodeficiency virus intermolecular triplex-forming oligonucleotide in anin vitrotranscription system due to RNase H-induced cleavage of the RNA transcript. Antisense Res Dev, 1993, 3(1):33–44.
[34]Papadakis G, Gizeli E.In silicosearch of DNA drugs targeting oncogenes. IEEE/ACM Trans Comput Biol Bioinform, 2012, 9(6): 1826–1830.
[35]Goni JR, de la Cruz X, Orozco M. Triplex-forming oligonucleotide target sequences in the human genome.Nucleic Acids Res, 2004, 32(1): 354–360.
[36]Morishita R, Higaki J, Tomita N, et al. Application of transcription factor “decoy” strategy as means of gene therapy and study of gene expression in cardiovascular disease. Circ Res, 1998, 82(10): 1023–1028.
[37]Morishita R, Aoki M, Kaneda Y. Decoy oligodeoxynucleotides as novel cardiovascular drugs for cardiovascular disease. Ann N Y Acad Sci, 2001, 947:294–301.
[38]Morishita R, Gibbons GH, Horiuchi M, et al. A gene therapy strategy using a transcription factor decoy of the E2F binding site inhibits smooth muscle proliferationin vivo. Proc Natl Acad Sci USA, 1995, 92(13):5855–5859.
[39]Zaug AJ, Been MD, Cech TR. The Tetrahymena ribozyme acts like an RNA restriction endonuclease.Nature, 1986, 324(6096): 429–433.
[40]Grosshans CA, Cech TR. A hammerhead ribozyme allows synthesis of a new form of the Tetrahymena ribozyme homogeneous in length with a 3′ end blocked for transesterification. Nucleic Acids Res, 1991, 19(14):3875–3880.
[41]Santoro SW, Joyce GF. A general purpose RNA-cleaving DNA enzyme. Proc Natl Acad Sci USA, 1997, 94(9):4262–4266.
[42]Kumar D, Chaudhury I, Kar P, et al. Site-specific cleavage of HCV genomic RNA and its cloned core and NS5B genes by DNAzyme. J Gastroenterol Hepatol,2009, 24(5): 872–878.
[43]Singh N, Ranjan A, Sur S, et al. Inhibition of HIV-1 integrase gene expression by 10-23 DNAzyme. J Biosci,2012, 37(3): 493–502.
[44]Fu S, Sun LQ. DNAzyme-based therapeutics for cancer treatment. Future Med Chem, 2015, 7(13): 1701–1707.
[45]Yu SH, Wang TH, Au LC. Specific repression of mutant K-RAS by 10-23 DNAzyme: sensitizing cancer cell to anti-cancer therapies.Biochem Biophys Res Commun,2009, 378(2): 230–234.
[46]Dass CR, Choong PF, Khachigian LM. DNAzyme technology and cancer therapy: cleave and let die. Mol Cancer Ther, 2008, 7(2): 243–251.
[47]Hong D, Kurzrock R, Kim Y, et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med, 2015,7(314): 314ra185.
[48]Duffy AG, Makarova-Rusher OV, Ulahannan SV, et al.Modulation of tumor eIF4E by antisense inhibition: a phase I/II translational clinical trial of ISIS 183750-an antisense oligonucleotide against eIF4E-in combination with irinotecan in solid tumors and irinotecan-refractory colorectal cancer. Int J Cancer, 2016, 139(7):1648–1657.
[49]Wang XZ, Wang SH, Song HF, et al. Pharmacokinetics of cantide, an antisense oligonucleotide, and its metabolites in rhesus monkeys. Acta Pharm Sin, 2011,46(11): 1370−1373 (in Chinese).王秀中, 王诗鸿, 宋海峰, 等. 反义寡核苷酸药物癌泰得及其代谢产物的猕猴体内药代动力学研究. 药学学报, 2011, 46(11): 1370–1373.
[50]Gilar M. Analysis and purification of synthetic oligonucleotides by reversed-phase high-performance liquid chromatography with photodiode array and mass spectrometry detection. Anal Biochem, 2001, 298(2):196–206.
[51]Mullis KB, Faloona FA. Specific synthesis of DNAin vitrovia a polymerase-catalyzed chain reaction.Methods Enzymol, 1987, 155: 335–350.
[52]Canene-Adams K. General PCR.Methods Enzymol,2013, 529: 291–298.
[53]Chandran S. Rapid Assembly of DNA via ligase cycling reaction (LCR). Methods Mol Biol, 2017, 1472:105–110.
[54]Kalin I, Shephard S, Candrian U. Evaluation of the ligase chain reaction (LCR) for the detection of point mutations. Mutat Res, 1992, 283(2): 119–123.
[55]Chen Y, Yang M, Xiang Y, et al. Ligase chain reaction amplification for sensitive electrochemiluminescent detection of single nucleotide polymorphisms. Anal Chim Acta, 2013, 796: 1–6.
[56]Walker GT, Fraiser MS, Schram JL, et al. Strand displacement amplification—an isothermal,in vitroDNA amplification technique. Nucleic Acids Res, 1992,20(7): 1691–1696.
[57]Wu W, Mao Y, Zhao S, et al. Strand displacement amplification for ultrasensitive detection of human pluripotent stem cells.Anal Chim Acta, 2015, 881:124–130.
[58]Detter JC, Jett JM, Lucas SM, et al. Isothermal strand-displacement amplification applications for high-throughput genomics. Genomics, 2002, 80(6):691–698.
[59]Demidov VV. Rolling-circle amplification in DNA diagnostics: the power of simplicity. Expert Rev Mol Diagn, 2002, 2(6): 542–548.
[60]Smolina IV, Demidov VV, Cantor CR, et al. Real-time monitoring of branched rolling-circle DNA amplification with peptide nucleic acid beacon. Anal Biochem, 2004, 335(2): 326–329.
[61]Kuhn H, Demidov VV, Frank-Kamenetskii MD.Frank-Kamenetskii, Rolling-circle amplification under topological constraints. Nucleic Acids Res, 2002, 30(2):574–580.
[62]Fujino M, Yoshida N, Yamaguchi S, et al. A simple method for the detection of measles virus genome by loop-mediated isothermal amplification (LAMP). J Med Virol, 2005, 76(3): 406–413.
[63]Notomi T, Mori Y, Tomita N, et al. Loop-mediated isothermal amplification (LAMP): principle, features,and future prospects. J Microbiol, 2015, 53(1): 1–5.
[64]Yang Z, McLendon C, Hutter D, et al.Helicase-dependent isothermal amplification of DNA and RNA by using self-avoiding molecularrecognition systems. Chembiochem, 2015, 16(9): 1365–1370.
[65]Vincent M, Xu Y, Kong H. Helicase-dependent isothermal DNA amplification. EMBO Rep, 2004, 5(8):795–800.
[66]Malek L, Sooknanan R, Compton J. Nucleic acid sequence-based amplification (NASBA). Methods Mol Biol, 1994, 28: 253–260.
[67]Compton J. Nucleic acid sequence-based amplification.Nature, 1991, 350(6313): 91–92.
[68]Van Ness J, Van Ness LK, Galas DJ. Isothermal reactions for the amplification of oligonucleotides. Proc Natl Acad Sci USA, 2003, 100(8): 4504–4509.
[69]Menova P, Hocek M. Preparation of short cytosine-modified oligonucleotides by nicking enzyme amplification reaction. Chem Commun (Camb), 2012,48(55): 6921–6923.
[70]Tan E, Wong J, Nguyen D, et al. Isothermal DNA amplification coupled with DNA nanosphere-based colorimetric detection. Anal Chem, 2005, 77(24):7984–7992.
[71]Lohmann JS, Stougaard M, Koch J. A new enzymatic route for production of long 5′-phosphorylated oligonucleotides using suicide cassettes and rolling circle DNA synthesis. BMC Biotechnol, 2007, 7: 49.
[72]Ducani C, Kaul C, Moche M, et al. Enzymatic production of ‘monoclonal stoichiometric’ single-stranded DNA oligonucleotides.Nat Methods, 2013, 10(7): 647–652.
[73]Wang X, Gou D, Xu SY. Polymerase-endonuclease amplification reaction (PEAR) for large-scale enzymatic production of antisense oligonucleotides. PLoS One,2010, 5(1): e8430.
[74]Li B, Dong S, Wu J, et al. Preparation of 5′-O-(1-Thiotriphosphate)-modified oligonucleotides using polymerase-endonuclease amplification reaction(PEAR). PLoS One, 2013, 8(7): e67558.
[75]Wang X, Gou D, Xu SY, et al. Enzymatic synthesis of modified oligonucleotides by PEAR using Phusion and KOD DNA polymerases.Nucleic Acid Ther, 2015, 25(1):27–34.
猜你喜欢
化工设计通讯(2022年6期)2023-01-02 22:40:32
中华诗词(2022年9期)2022-07-29 08:33:50
中国慈善家(2022年3期)2022-06-14 22:21:55
快乐语文(2021年34期)2022-01-18 06:04:14
小学生学习指导·低年级(2021年6期)2021-09-10 14:07:49
考试与评价·七年级版(2021年4期)2021-08-14 10:45:54
中国(俄文)(2020年8期)2020-11-23 03:37:13
小学阅读指南·低年级版(2018年5期)2018-11-02 10:19:50
中国生化药物杂志(2015年7期)2015-07-07 15:15:58
生物技术世界(2014年7期)2014-08-15 00:43:39
