
一种提高变体库多样性的寡核苷酸设计方法
2022-08-18 02:16唐瑞郑浩然
北京生物医学工程 2022年4期
唐瑞 郑浩然
0 引言
目前合成生物领域已有许多可以获得具有各种改进酶特性的蛋白质的方法。相较于传统方法对蛋白质进行理性设计,定向进化[1]因为不需要具有关于酶的结构和机制等先验知识而被更广泛地使用。该方法通过诱变或基因重组来随机生成大型基因变体库(library of variants),然后通过高通量筛选得到具有改进特性的变体基因[2-3]。目前已经有许多策略用于构建基因变体库,例如易错聚合酶链式反应(error prone polymerase chain reaction,epPCR)介导的随机诱变[4-5]、DNA改组[6](DNA shuffling)以及目标位置的饱和诱变[2]等。
其中DNA改组是一种强大的技术,可以开发用于蛋白质定向进化的基因变体库[6]。在该方法中,通过基于同源或非同源的重组技术从单个基因或基因家族中得到随机片段产生多种变体[7]。但是传统的DNA改组有许多缺点。一是在彼此差异较大的变体序列之间改组的可能性很低[8]。即使彼此非常接近的突变体改组也可能效率低下[9]。二是从更多不同的序列中创建的变体库大多倾向于产生亲本同源双链体,嵌合体中的交叉互换事件发生的数量非常低[2]。为了克服上述缺陷,目前已经开发了许多改进的方法。其中一种简单的并且可以减少具有高度同源序列区域偏差的方法就是从头寡核苷酸合成策略[9]。
从头寡核苷酸合成策略包含多种具体实现方法。例如合成改组(synthetic shuffling)通过重叠延伸聚合酶链式反应组装简并寡核苷酸以构建包含不同序列的变体库[10]。基因组装诱变(gene assembly mutagenesis)使用简并寡核苷酸来保证目标位置的饱和诱变并通过基因组装获得变体库[9,11]。当模板序列难以随机化时,基于设计的寡核苷酸组装(assembly of the designed oligos,ADO)重组可以构建更高质量的基因变体库[9]。此外,使用这些基于寡核苷酸合成的方法有几个特殊的优势。一是不需要基因的DNA。其次,在设计和合成阶段,亲本基因的自杂交被最小化或消除[12]。
尽管涉及从头寡核苷酸合成的这些方法比经典改组方法具有许多优势,但由于简并合成寡核苷酸及其数量等原因,这些方法的好处并未完全体现。究其深层次原因之一是合成寡核苷酸的高成本。但是随着技术的发展,使用基因芯片合成技术可以降低寡核苷酸的合成成本[13]。此外,利用基因芯片合成寡核苷酸进行基因组装的发展[13-16]也使得ADO方法能够用于构建高多样性的基因变体库[9]。
在过去十年中,基于基因芯片的从头寡核苷酸合成取得了重大进展[13-16]。因此,ADO与大规模合成寡核苷酸的结合成为可能。但同时,对这些大量的将被合成并用于变体库构建的寡核苷酸的设计算法就变得尤为重要。
目前已经研究开发出了许多用于从头合成的寡核苷酸设计工具。其中包括 DNAWorks[17]和TmPrime[18]。然而,由于这些软件工具设计之初的应用对象是传统的DNA序列合成实验,因此考虑到序列间的同源区域,这些软件程序不支持嵌合寡核苷酸的生成。相反,当输入多条同源序列时,大多数软件会努力避免寡核苷酸之间的相似性。因此,在基于基因芯片合成技术的从头寡核苷酸合成上,需要一个新的寡核苷酸设计方法和工具。为此课题组设计了一款HomoLib(homologous library)的工具,该工具能够为了得到定向进化所需的高多样性基因变体库而进行寡核苷酸设计。
1 方法
HomoLib包括3个功能模块,分别是基因设计模块、基因切片模块和寡核苷酸设计模块,通过协同工作设计满足定向进化要求的高多样性的基因变体库所需的寡核苷酸,同时这3个模块也可以独立工作,用作其他领域。
1.1 基因设计模块
基因设计模块的目的是获得亲本氨基酸序列对应的DNA序列。HomoLib从比对的蛋白质序列中设计DNA序列,旨在以最大限度提高不同DNA序列之间的一致性。该模块包括多序列比对(multiple sequence alignment,MSA)程序、逆翻译程序、密码子优化程序和引物添加程序(图1)。
在输入亲本氨基酸模板序列后,所有3个独立模块的第一步都是进行输入序列的比对(图1)。MSA采用渐进式技术的启发式搜索,该技术于1984年开发[19],也被称为分层或树方法。渐进比对通过将两两比对迭代应用于最相似的序列对来构建最终的MSA(图2)。然后比对的氨基酸序列被逆翻译成DNA序列。逆翻译程序通过最大化不同序列间的一致性来设计DNA序列。

图2 多序列比对算法示意图Figure 2 Diagram of multiple sequence alignment algorithm
基因设计模块的目的是为了使DNA序列尽可能同源,即通过选择密码子来提高DNA序列的相似性。为了实现这一点,HomoLib使用候选密码子的组合来创建给定位置的候选密码子库,对于每个密码子组合,计算平均相似度分数并选择具有最高相似度分数的最佳密码子组合。该程序的流程在图3中进行了说明。

图3 逆翻译算法示例Figure 3 Example of reverse translation algorithm
为了避免依次删除限制位点可能会重新引入限制位点的情况,HomoLib采取全局策略来同时删除所有限制位点。首先定义了一个新名词叫位点簇子序列(site-cluster subsequence),其中限制位点的任何密码子替换都不会将新的限制性位点引入相应序列的其余部分,因此对位点簇的密码子替换不会在其他位置引入限制位点。首先,密码子优化程序会找到每个DNA序列的所有位点簇子序列。其次,对于每个位点簇子序列,将尝试用所有候选密码子替换原始密码子,用以消除限制位点。最后,对于使位点簇子序列无限制位点的密码子(通常有多个),选择与原始密码子相比核苷酸变化最少的作为最佳密码子,目的是为了保持较高的相似性。
1.2 基因切片模块
从基因设计模块获得DNA序列后,可以视序列长度来决定是否进行基因切片,目的是将过长且难以组装的DNA序列分成几个较短的片段(图1)。经过基因切片后,后续在全长DNA组装期间将会提供在片段之间改组的机会,从而提高最终变体库的多样性。为了提高改组的重组率,应在DNA序列的高度同源区域切割片段。不同片段之间的受控重叠区域可用作同源重组中的连接段(linker)。该模块的算法实现包括两个步骤:根据片段长度范围和全长比对的DNA序列来初始化切割位点,然后微调初始化的切割位点以生成具有高度同源区域的最佳切割位点。
1.3 寡核苷酸设计模块
在寡核苷酸设计模块中,将对短DNA序列切割成用于DNA合成需要的寡核苷酸。为了提高寡核苷酸合成的效率,需要限制寡核苷酸的长度。该模块确保相邻的寡核苷酸之间没有间隙,从而降低了DNA组装过程中的难度和错误率。该模块的主要步骤如下。
(1) 在寡核苷酸长度范围的约束下,从5’(five prime)端开始到3’(three prime)端结束,一一识别具有高同源区域的正向切割位点。这样设计的寡核苷酸将在对应的DNA序列区域具有相似的5’端和(或)3’末端。
(2) 在寡核苷酸长度范围的约束下,从5’端开始到3’端结束,一一识别具有高同源区域的反向切割位点,并且每个反向切割点应位于两个相邻的正向切割点之间。
由于寡核苷酸的长度比较短小,因此不可能每个寡核苷酸的两端都具有较高的同源性,于是HomoLib提供了2个选项供用户选择。高同源序列可以选择5’端和3’端都具有较高的同源性,默认只要求3’端具有较高同源性。因此,对于高同源DNA序列的顶部链(top strand),该算法在MSA之后寻找高度同源区域中间的切割点。相应反向寡核苷酸的末端移动到底部链(bottom strand)的相邻同源区域的中间。
2 结果与分析
2.1 基因序列的一致性
21个红色荧光蛋白(red fluorescent protein,RFP)的氨基酸序列被输入到基因设计模块中用以得到基因序列,使用的宿主细胞是大肠杆菌。
基因设计模块可以将氨基酸序列逆向翻译成相似度更高的DNA序列。图4提供了HomoLib、DNAWorks和TmPrime之间逆翻译DNA序列的一致性比较,其中一致性的计算是使用的Clustal Omega软件。本文方法优化了逆翻译过程中密码子的选择。然而,DNAWorks和TmPrime为了规避交叉匹配的情况不会考虑提高基因序列的一致性。因此,与使用DNAWorks和TmPrime逆翻译的DNA序列相比,使用HomoLib逆翻译的DNA序列一致性是最高的。通过我们的方法,逆翻译得到的DNA序列的平均一致性为84.41%。该值高于亲本氨基酸序列(平均一致性69.82%),而DNAWorks和TmPrime的平均一致性分别仅为69.58%和67.56%。由于一致性70%以上时重组更为有效[20],从结果中可以看出HomoLib得到的逆翻译DNA序列将使得重组效率更高。
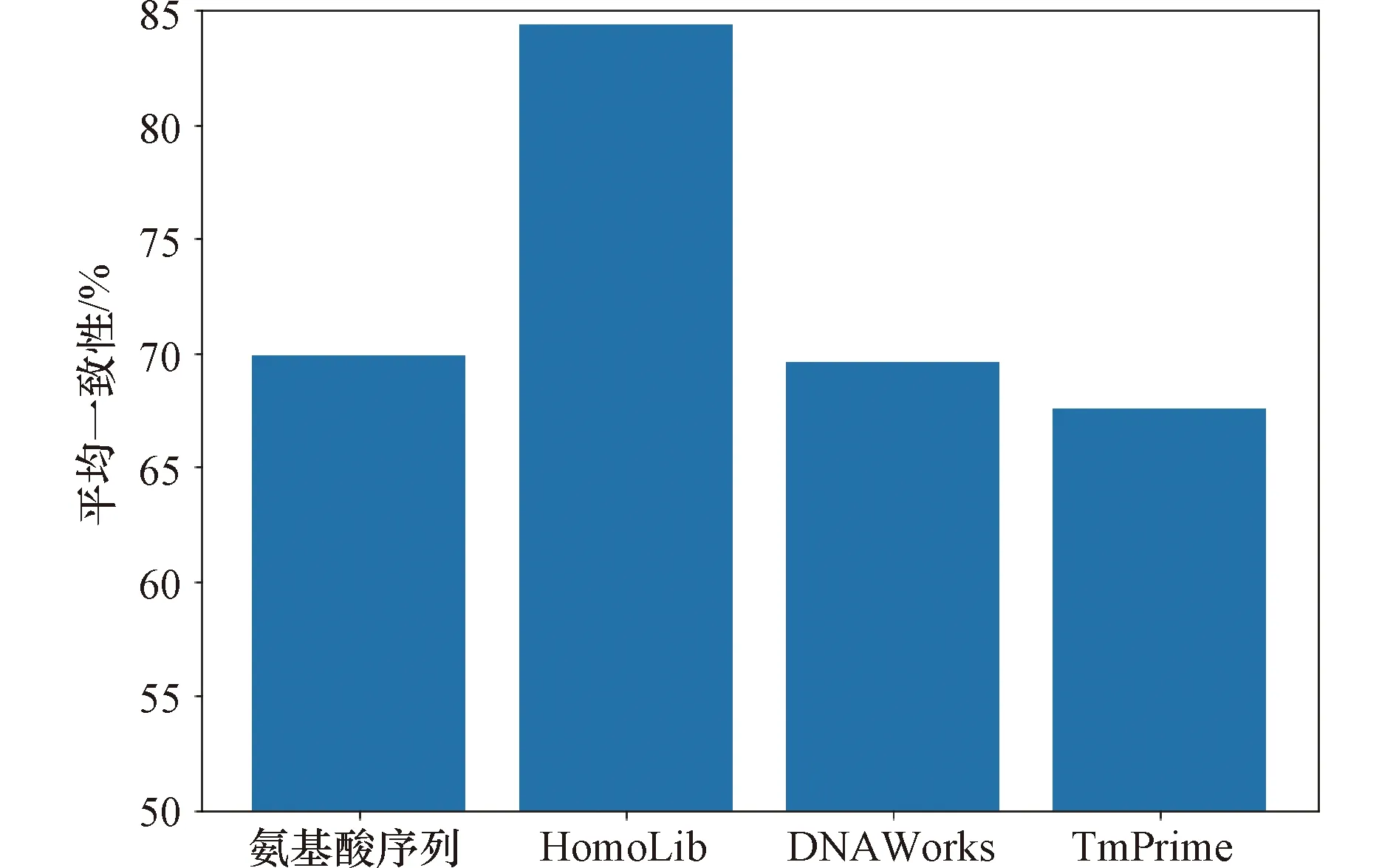
图4 序列间的平均一致性Figure 4 Average identity between sequences
2.2 基因合成过程中的重组率
基因切片模块可以将全长序列切分成具有相似末端的片段。因此,在将片段组装成全长 DNA 的过程中,发生重组的可能性将得以增加。如图5所示,DNA序列在高同源区被切割成片段,在末端有长达28 bp(base pair)的重叠,该重叠长度可由用户自定义设置。切割位点在高同源区域,确保不同基因序列的所有末端都尽可能相似。

图5 DNA切割片段末端的重叠区域Figure 5 Overlapping region at the ends of DNA fragments
2.3 寡核苷酸末端的高相似性
由于长度是影响寡核苷酸Tm(melting temperature)值的重要参数,因此在寡核苷酸设计中,考虑到序列的相似性,通过限制相似区域的长度,将同源区域的Tm值限制在湿实验的要求范围内。在将21个RFP的DNA序列切割成具有重叠末端的短片段后,使用寡核苷酸设计模块将片段进一步切割成寡核苷酸。课题组比较了HomoLib以及DNAWorks和TmPrime的这些关于将DNA序列切割成寡核苷酸的数据结果,本文方法根据序列的同源区域确定了切割位点,切割后每条链的末端显示出明显的相似性,提高了重组效率。但是DNAWorks和TmPrime没有考虑序列同源性,切割后末端高度相似不明显。
3 讨论
蛋白质定向进化流程中,许多合成生物技术都可以生成基因变体库,为了克服传统基因变体库生成方式的诸多缺点,研究人员采用了基于基因芯片技术的从头寡核苷酸合成方法来生成基因变体库,并提高其多样性。由于目前广泛使用的寡核苷酸设计工具是基于传统的DNA合成技术而设计的,有避免寡核苷酸或基因间相似的特性,而这阻碍了基于基因芯片技术的从头寡核苷酸合成基因变体库的多样性。因此,本文提出了HomoLib工具,其可以对蛋白质合成实验进行寡核苷酸设计,并以此提高合成后基因变体库的多样性。
本文提出的方法主要适用于采用从头寡核苷酸合成策略的基因芯片技术的定向进化。在后续的研究中还需要对其他生物合成方法进行改进以及针对低同源序列进行优化。
4 结论
本文提出的HomoLib工具,可以针对输入氨基酸序列进行密码子优化、基因切片等处理,从而提高序列间的一致性,进而增加基因变体库合成过程中的重组率,最后提高变体库的多样性。逆翻译后的基因序列一致性的提升、高同源区域的切割以及后续基因变体库的成功合成及筛选证实了该工具的有效性。
猜你喜欢
今日农业(2022年1期)2022-11-16
今日农业(2022年2期)2022-11-16
分子催化(2022年1期)2022-11-02
中国农业科学(2022年16期)2022-09-19
电脑报(2022年11期)2022-06-14
北京航空航天大学学报(2022年5期)2022-06-06
作物学报(2022年3期)2022-01-22
电脑知识与技术(2018年19期)2018-11-01
智富时代(2018年10期)2018-01-30
智富时代(2018年10期)2018-01-30
