
15N标记L-胱氨酸
2010-07-18 01:25:56汤财德李美华宋家龙陆平晔杜晓宁
同位素 2010年2期
罗 勇,汤财德,李美华,宋家龙,陆平晔,杜晓宁
(上海化工研究院上海稳定同位素工程技术研究中心,上海 200062)
15N是氮元素的稳定同位素之一,可以用作示踪元素,广泛应用于生物化学、医学、药物学、农业科学等领域。15N标记氨基酸和多肽是研究医学和生命科学的重要工具,在蛋白质的人工合成、判定生物活性物质(多肽、蛋白质)的化学结构及药用机理等方面起着独特的示踪作用,其应用越来越广泛,市场前景良好[1]。L-胱氨酸和L-半胱氨酸均为含硫氨基酸,属人体非必需氨基酸,在体内均可由蛋氨酸转变而来。其中,从L-半胱氨酸出发可生成谷胱甘肽(G-SH),由于其活性基团巯基的存在,使之具有许多重要的生理功能,如整合解毒作用、增强肝功能、抗氧化作用和促进毛发生长等。L-胱氨酸同时也是多种硬蛋白质如角质蛋白的重要成分。
L-胱氨酸广泛存在于动物的毛、发、骨、角中,目前工业上主要采用生物化学方法,经由蛋白质(如毛发)水解、分离、精制而成。L-胱氨酸的化学生产具有较大的难度,合成过程中需对巯基进行保护和解保护[1-2]。此外,经化学方法合成的通常为消旋的D,L-氨基酸,不具有光学活性,必须经化学或酶法拆分以得到有光学活性的L型产物。
15N标记的L-胱氨酸是研究蛋白质代谢的重要示踪剂之一,目前国际上仅ISOTOPE等少数公司能提供产品,价格昂贵,售价高达每克4 640美元。因此开发15N标记L-胱氨酸的制备工艺具有较好的学术意义和经济效益。
基于L-胱氨酸可在酸性溶液中还原为L-半胱氨酸,L-半胱氨酸也可在碱性环境中氧化为L-胱氨酸,本实验拟设计一个新的合成路线合成15N-L-胱氨酸。元素分析仪:美国PE公司;LC-20AT高效液相色谱:日本岛津公司;Thermo Finingan TSQ/A ccela质谱仪:美国热电公司;MAT-271气体同位素质谱计:西德菲尼根玛特公司。
15NH4Cl:上海化工研究院生产;邻苯二甲酸、丙二酸二甲酯、苄硫醇、三聚甲醛等皆为市售化学纯或分析纯试剂。
2 实验方法
总体合成路线设计为三步:第一步先合成S-苄基-D,L-半胱氨酸;第二步,采用酶法拆分S-苄基-D,L-半胱氨酸,包括对 S-苄基-D,L-半胱氨酸乙酰化、酶拆分和消旋化;第三步,对拆分后的S-苄基-D,L-半胱氨酸解保护,脱除苄基后再经氧化得到L-胱氨酸。
2.1 S-苄基-D,L-半胱氨酸的合成
1 主要仪器与试剂
WRS-1数字熔点仪:上海精密仪器有限公司;Bruker biospin AC-P200型核磁共振仪:德国Bruker公司;Nicolet FT-IR 6770红外吸收光谱仪:美国Nicolet公司;Perkin-Elmer 240C
S-苄基-D,L-半胱氨酸的合成采用经过改进的Gabriel方法,其合成路线示于图1。邻苯二甲酸钾盐(1)与溴代丙二酸二甲酯缩合得到邻苯二甲酰亚胺抱丙二酸二乙酯(2),其钠盐(3)再与苄基氯甲硫醚(4)反应得到N-苄基氯甲硫醚基邻苯二甲酰亚胺抱丙二酸二乙酯(5),再经水解得到S-苄基-D,L-半胱氨酸(6)。

图1 S-苄基-D,L-半胱氨酸的合成路线
2.1.1 邻苯二甲酰亚胺钾盐(1)的制备
邻苯二甲酰亚胺钾盐(1)的合成参考文献[3-4]。将NaOH溶液缓慢滴加到26.75 g(0.50mol)NH 4 Cl中,生成的 NH 3随 N2气流通入由83.1 g(0.50 mo l)邻苯二甲酸和200m L水配成的悬浊液中。待NH 3完全被邻苯二甲酸溶液吸收后,先于100℃下蒸除溶液中的水,然后缓慢升温至200℃脱水,最后将温度逐渐上升至300℃,冷却得白色固体74.1 g。将上述制得的邻苯二甲酰亚胺粗品溶于无水乙醇,于搅拌下缓慢滴加KOH的无水甲醇溶液,室温下保持3 h,过滤,用无水乙醇洗涤3次,得邻苯二甲酰亚胺钾盐(1)白色晶体88.9 g,收率96.1%。
2.1.2 邻苯二甲酰亚胺抱丙二酸二乙酯(2)及其钠盐(3)的制备
将18.52 g(0.1 mol)N-邻苯二甲酰亚胺钾盐(1)与23.90 g(0.1m ol)新鲜制备的溴代丙二酸二乙酯均匀混合,在110℃油浴中加热1 h,冷却后加水搅拌,反应物变为固体。将其转移到研钵内反复研磨,水洗抽滤后得到29.80 g(2),收率97.6%。将制得的产物(2)加热溶解于100 m L甲苯中,加入2.75 g(0.12 mol)金属钠条,回流搅拌 3 h,沉淀经甲苯洗涤,干燥,得30.7 g黄色产物(3),收率96.2%。
2.1.3 苄基氯甲硫醚(4)的制备
将49.7 g(0.40 mol)苄硫醇与40.5 g(0.45m ol)三聚甲醛在冰浴中冷却,并用干燥过的HCl气饱和,然后加入 70 g(0.63 m ol)CaCl2,再于室温下保持24 h。过滤除去固体物质后,母液用5 mol/L NaOH溶液洗涤3次,无水Na2 SO4干燥,过滤后,减压(0.27 kPa)蒸馏,收集 102℃的馏分,得到苄基氯甲硫醚(4)21.1 g,收率30.5%。
2.1.4 N-苄基氯甲硫醚基邻苯二甲酰亚胺抱丙二酸二乙酯(5)的制备
将5.00 g(0.015 3mol)钠盐(3)和2.70 g(0.015 6 mo l)苄基氯甲硫醚(4)和50 m L无水甲苯搅拌回流12 h,过滤。沉淀经甲苯洗涤后,再用无水乙醇重结晶,得到产物(5)4.0 g,产率59.4%。产物(5)的熔点为79.5~80.4℃;1H NMR(CDCl3):1.28(t,6H,J=7.0 H z,—CH 3),3.55(s,2H, —C—CH 2—S—),3.70(s,2H,—S—CH2—A r),4.32(q,4H,J=7.0Hz,—OCH2—),7.10 ~ 7.27(m,5H,—C6H5),7.72~7.81(m,4H,—C6H4)。
2.1.5 S-苄基-D,L-半胱氨酸(6)的制备
将4.0 g缩合产物(5)悬浮于25m L V(乙醇)∶V(水)=1∶1混合液中,于搅拌下逐滴加入20m L 5 mol/L NaOH,保持温度55~60℃,搅拌1 h后,加入足量浓盐酸酸化,加热水解2 h后蒸干溶液,滴加少许NaOH溶液至溶液呈中性(刚果红)。将得到的沉淀过滤,并悬浮于沸腾的95%乙醇中,趁热过滤,冷却后得到晶体状S-苄基-D,L-半胱氨酸(6)0.98 g,收率50.1%。产物(6)的熔点:211~214℃;元素分析实测值(计算值,%):C 56.80(56.87),N 6.58(6.63),H 6.15(6.20);MS-EI(m/z,%):211.1(M+,10),91.1(100);IR(υ/cm-1,KBr):3 062(C —H,A r—H),2 934(O—H,—COOH),1 500,1 588(C —C,benzene),1 400(C=O,—COOH)。
2.2 氨基酰化酶法拆分S-苄基-D,L-半胱氨酸
氨基酰化酶法拆分S-苄基-D,L-半胱氨酸的化学过程示于图2。
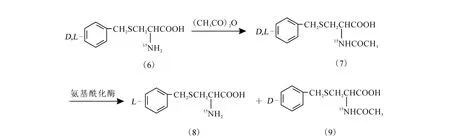
图2 氨基酰化酶法拆分S-苄基-D,L-半胱氨酸的化学过程
2.2.1 N-乙酰-S-苄基-D,L-半胱氨酸的(7)的制备
将6.0 g(0.028 4 mol)D,L-硫苄基半胱氨酸(6)溶于48m L 1 mol/LNaOH溶液中,于冰浴下逐滴滴加12 m L乙酸酐,保持2 h后,再用浓盐酸酸化至pH 为2,经抽滤、水洗、干燥,得到6.90 g白色固体(7),收率95.9%。熔点:158.7℃。
2.2.2 N-乙酰-S-苄基-D,L-半胱氨酸的拆分
[5-8]对产物(7)进行拆分:将6.90 g(0.027 2m ol)上述制得的 N-乙酰-S-苄基-D,L-半胱氨酸溶于50 m L 1 mol/L NaOH溶液中,调节溶液的pH 约为8.0,将温度控制在37℃,加入0.20 g氨基酰化酶,于轻微搅拌下保持30 h,再调节溶液的pH至5.5,4℃过夜,抽滤,洗涤后得到白色固体S-苄基-L-半胱氨酸(8),收率 82.6%。熔点:213.6℃,比旋光[α]D=+19.4°(NaOH 浓度为1mol/L,20 ℃)。
2.2.3 N-乙酰-S-苄基-D-半胱氨酸(8)的消旋
参考文献[9]进行消旋化:称取3.0 g产物(8)溶于20 m L 2.5 m ol/L NaOH中,升温至60~65℃,保温1 h。逐滴加入20 m L乙酸酐,恒温30m in,冷却。用浓盐酸将溶液酸化至pH=2,保持4℃过夜,沉淀物过滤烘干后用于二次拆分,可继续得到白色固体(8)。经二次拆分后,S-苄基-L-半胱氨酸(9)的拆分总收率为62%。
2.3 15N-L-胱氨酸(11)的制备
对N-乙酰-S-苄基-D,L-半胱氨酸(9)进行解保护/氧化,制备L-胱氨酸,其化学过程示于图3。
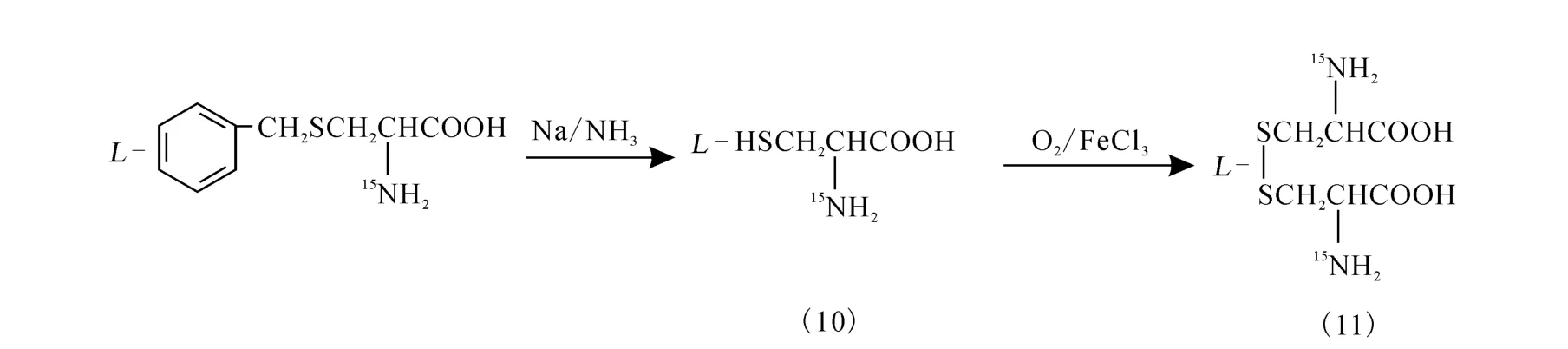
图3 对N-乙酰-S-苄基-L-半胱氨酸解保护/氧化得到L-胱氨酸的化学过程
在-50 ℃下,将 3.5 g(0.016 5 m ol)产物(9)加入到50m L液氨中,再缓慢加入小块金属钠,维持蓝色3~5 min不褪,空气中蒸发除去液氨,加入50 m L水;调pH 至8,加入痕量FeCl3,通入空气流氧化;经亚硝酸铁氰化钠检测确定无硫醚后,过滤干燥。得白色固体(11)1.29 g,收率:78.0%。熔点:258℃。
2.4 15N-L-胱氨酸的丰度实验
在上述实验基础上,以15NH 4 Cl(10.20%15N)为原料,进行L-胱氨酸合成的丰度验证实验,具体合成方法如前所述。
3 结果与讨论
由于稳定同位素15N原料非常昂贵,衡量合成路线优劣的主要因素是稳定同位素原子的利用率、产物的生物活性和同位素丰度。其中,经Gabriel法改良的丙二酸酯法是15N标记氨基酸的重要合成方法。L-胱氨酸的巯基非常活泼,在合成过程中需对此加以保护。可供选择的保护基有苄基、二苯甲基、三苯甲基等,其中苄基最为常用。因此,本工作采用苄基作为巯基保护基,将商业上较易获得的苄硫醇引入,制得苄基氯甲硫醚,并作为合成前体。此外,经化学合成方法得到的通常为消旋物,不具有光学活性,因此必须经化学或酶法拆分得到L型产物,最后,再经解保护过程脱除保护基,得到目标产物15N标记L-胱氨酸。
3.1 S-苄基-D,L-半胱氨酸合成中的影响因素
在15N-L-胱氨酸的合成过程中存在多个关键合成步骤,对最终产物的收率影响很大。
3.1.1 反应温度对邻苯二甲酰亚胺抱丙二酸二乙酯(2)收率的影响
在产物(2)的合成过程中,保持其他条件不变,仅改变油浴温度,观察温度对产物(2)收率的影响,结果列于表1。由表1可知,在实验条件下,反应温度对邻苯二甲酰亚胺抱丙二酸二乙酯的收率有较大影响,在110~120℃温度范围内可获得良好的收率。随着温度进一步升高(高于130℃),产物的熔程变宽,表明副产物增多,收率降低。
3.1.2 反应时间对N-苄基氯甲硫醚基邻苯二甲酰亚胺抱丙二酸二乙酯(5)收率的影响
产物(5)的合成过程中,保持其他条件不变,仅改变反应时间,观察反应时间对产物(5)收率的影响,结果列于表2。由表2可知,在实验条件下,反应时间对邻苯二甲酰亚胺抱丙二酸二乙酯的收率有较大影响,随着反应时间延长,产物收率显著增加;12 h以后,进一步延长反应时间(16 h),反应收率不再有明显变化,因此,该步反应时间以12 h为宜。
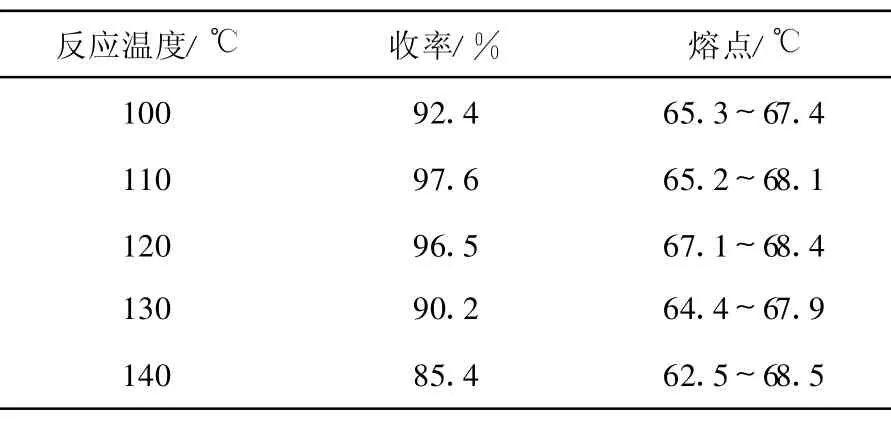
表1 反应温度对邻苯二甲酰亚胺抱丙二酸二乙酯(2)收率的影响
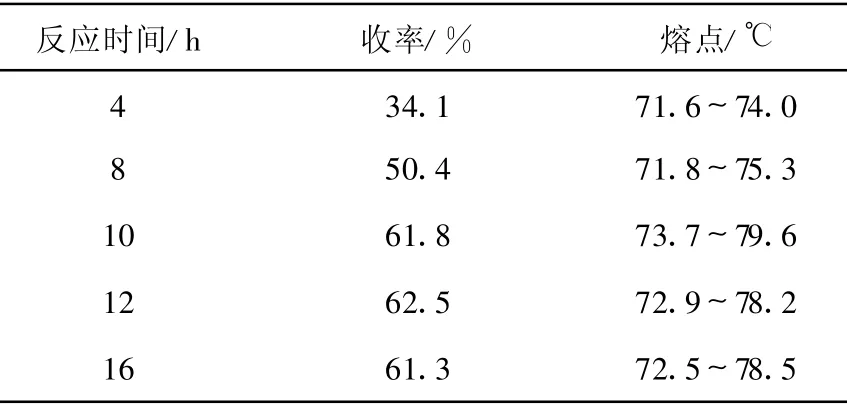
表2 反应时间对N-苄基氯甲硫醚基邻苯二甲酰亚胺抱丙二酸二乙酯(5)收率的影响
3.1.3 水解时间对S-苄基-D,L-半胱氨酸(6)收率的影响
在探索实验中发现,N-苄基氯甲硫醚基邻苯二甲酰亚胺抱丙二酸二乙酯的水解时间是影响S-苄基-D,L-半胱氨酸收率的重要因素。反应按2.1.5进行,在其他条件不变的情况下,只改变盐酸水解的时间,考察水解时间对D,L-硫苄基半胱氨酸收率的影响,结果列于表3。由表3可知,随着反应时间延长,产品收率上升,2 h时收率最高,达50%,此后随着水解时间进一步延长,产品收率略有下降。因此,实际水解时间应控制在2 h。

表3 水解时间对 S-苄基-D,L-半胱氨酸收率的影响
3.2 氨基酰化酶法拆分D,L-硫苄基半胱氨酸中的影响因素
将所制得的S-苄基-D,L-半胱氨酸进行乙酰化,并采用氨基酰化酶对其进行光学拆分,再将具有光学活性的S-苄基-L-半胱氨酸从拆分混合液中分离出来。为了提高拆分的收率,还需对未参与反应的N-乙酰-S-苄基-D-半胱氨酸进行消旋化,进行再次拆分。
在本研究过程中,先后通过单因素考察和正交实验,对影响氨基酰化酶法拆分N-乙酰-S-苄基-D,L-半胱氨酸生成S-苄基-L-半胱氨酸的多种影响因素进行了优化,找到了比较合适的工艺条件[11]:反应物 N-乙酰-S-苄基-D,L-半胱氨酸的量为5 g,pH为8.0,反应温度为37℃,酶用量为200 m g,反应液总体积为50 m L,反应时间为30 h。在此条件下,S-苄基-L-半胱氨酸拆分的单程收率达到45%,N-乙酰-S-苄基-D-半胱氨酸回收率达到88%。经过两次拆分以后,S-苄基-D,L-半胱氨酸总的拆分收率达到62%。
3.3 保护基的脱除
解保护即脱除S-苄基-L-半胱氨酸上的保护基苄基。可供选择的体系有HF体系[10]和Na/NH3体系。HF体系收率较高(90%),但HF气体毒性大,对于实验装置要求很高;Na/NH3体系收率相对较低(78%),操作相对容易,危险性较小。因此在实验中选择Na/NH3体系进行解保护。由于L-半胱氨酸在中性和碱性溶液中易被氧化生成 L-胱氨酸,且 L-胱氨酸难溶于水,因此在解保护后,在痕量Fe3+的催化下,于近中性溶液中将L-半胱氨酸氧化为L-胱氨酸。
3.4 产品的分析与表征
对于终产品进行红外分析,结果示于图4。根据图 4可计算 IR(υ/cm-1,KBr)得:2 980(O —H,—COOH),1 680(C=O,—COOH),1 590(N—H,—NH2),1 480(C—H,—NH2—),1 400(C—O,—COOH),1 110(C—N,—CH—NH2)。薄层层析鉴定表明,产物与L-胱氨酸标样具有相同的R f。比旋光度[α]D为-215°(c=1,1 m ol/L HCl,20 ℃),与标样在该条件下的比旋光度-220°很接近。在HPLC上检测产物纯度大于98%。元素分析,C6H12N2S2O4实测值(计算值):C 29.82%(29.99%),N 11.59%(11.66%),H 6.15%(6.13%)。

图4 L-胱氨酸的红外光谱
3.5 15N-L-胱氨酸丰度实验验证
通过优化合成方法,以NH 4Cl和苄基氯甲硫醚为前体进行了L-胱氨酸的合成,总收率为13.9%,产物的化学纯度和光学纯度符合要求。在此基础上,以15NH4Cl(10.20%15N)为标记前体,进行L-胱氨酸合成的丰度实验,合成总收率13.5%,15N的丰度为10.18%,未出现同位素稀释现象。
4 小 结
设计并完成了适合15N标记的L-胱氨酸,以15NH4 Cl为起始原料,通过改进的Gabriel方法得到S-苄基-D,L-半胱氨酸,再经氨基酰化酶法拆分和解除保护后得到15N标记L-胱氨酸。红外、HPLC质谱、元素分析等分析手段均确证了多种中间体和15N标记L-胱氨酸的结构;以同位素原料计总产率为13.5%,所用工艺不会产生同位素稀释,可以满足商品化15N标记L-胱氨酸及相关衍生物的合成。
参考文献:
[1] John W,Vincent V.A new synthesis of cystine[J].JBio Chem,1939,131(1):267-271.
[2] Masatsune Kenichi U.Synthesis o f[1,1'-13C2]-L-cysteine[J].J Label Comp Radiopharm,1991,29(8):867-874.
[3] 盛怀禹,陈耀焕,袁群,等.同位素有机化学[M].浙江:浙江教育出版社,1994:209-214.
[4] Authur M,L loyd W.Organic syntheses w ith isotopes:partⅡ[M].New York:Interscience publishers,1958:1 776-1 788.
[5] Yuan YJ,Wang SH,Song ZX,et al.Production of L-methionine by immobilized pellets of Aspergillus oryzae in a packed bed reactor[J].JChem Tech Bio,2002,77(6):602-606.
[6] 张倩颖,张关永.S-苄基-L-半胱氨酸测定方法的研究[J].氨基酸和生物资源,2000,22(3):62-64.
[7] 姚文兵,侯振清,吴梧桐,等.固定化牛肾氨基酰化酶拆分法制备D-丙氨酸[J].中国药科大学学报,2000,31(4):297-300.
[8] 张清玉,谭欣,赵林,等.乙酰-D-氨基酸消旋工艺的研究[J].化学工业与工程,2004,21(2):91-95.
[9] John W,Vincent V.Racem ization of benzy l-L-cysteine,with a new method of p reparing D-cystine[J].JBio Chem,1939,130(1):109-114.
[10]Shumpei S,Yasutsugu S,Yasuo K,et al.Use of anhydrous hydrogen fluoride in peptide synthesis:I.Behavior of various perotective groups in anhydrous hydrpgen fluoride[J].Bu ll Chem Soc Japan,1967,40(9):2 164-2 167.
[11] 柯昌武,罗勇,潘洁,等.酶法拆分15N标记 S-苄基-半胱氨酸的研究[J].上海化工,2009,34(10):7-11.
猜你喜欢
山西化工(2023年10期)2023-11-15 08:47:42
探索科学(学术版)(2020年4期)2021-01-18 02:28:56
环境污染与防治(2021年11期)2021-01-09 02:08:51
临床儿科杂志(2020年2期)2020-03-12 04:04:10
发明与创新(2019年42期)2019-11-18 01:24:04
百科知识(2016年18期)2016-10-28 00:20:12
山东化工(2016年4期)2016-09-05 12:30:54
灾害医学与救援(电子版)(2016年2期)2016-03-11 20:18:04
医学研究杂志(2015年3期)2015-06-10 06:41:52
中国医疗美容(2015年4期)2015-04-27 02:24:11
