
机械通气肺损伤时肾素-血管紧张素系统激活的实验研究*
2010-05-16 02:04朱宏飞姚尚龙武庆平
华中科技大学学报(医学版) 2010年2期
朱宏飞 冯 丹 姚尚龙 武庆平
华中科技大学同济医学院附属协和医院麻醉科,武汉 430022
肾素-血管紧张素系统(RAS)不只存在于心血管系统,研究表明,在人和大鼠肺组织中,也存在RAS,并对肺组织细胞的生长、凋亡起着重要的调节作用,其中尤以血管紧张素Ⅱ(ANG Ⅱ)及其1型受体(AT1)较为关键[1]。近些年来研究发现,肺损伤时肺组织ANGⅡ的含量显著增加,而减少ANG Ⅱ的生成或阻断AT1可以减轻多种原因所致的肺损伤[2-4];Marshall等[5]的一项大规模前瞻性研究也表明,血管紧张素转化酶(ACE)抑制剂的活性关系到ARDS的发生、发展及预后,提示RAS可能在急性肺损伤的致病机制中起着非常重要的作用。目前,有关RAS在机械通气肺损伤(ventilator-induced lung injury,VILI)中的作用还少见报道。本实验主要研究大鼠VILI时RAS的激活情况。
1 材料与方法
1.1 主要仪器和试剂
小动物呼吸机(哈佛683,美国);血气分析仪(i-State,美国);石蜡切片机(Leika超薄切片机,德国);PCR 仪(Bio-Rad MJ PTC-100TM,美国);半干式转膜仪(Bio-Rad 170-3940,美国);酶标仪(Biorad 680,美国)。T rizol提取液、RT-PCR试剂盒及ANG ⅡELISA试剂盒(深圳晶美公司);总蛋白和MPO检测试剂盒(南京建成生物工程研究所);ACE一抗(Santa Cruz,美国);ECL检测试剂盒(Amersham,美国)。
1.2 动物与分组
华中科技大学同济医学院实验动物学部动物实验中心内进行实验,中心合格证号:SYXK(鄂)2004-0028。24只健康雄性SD大鼠(同济医学院实验动物学部提供),体重300~350 g,随机分成4组(每组6只大鼠):A为空白对照组,不行机械通气;B、C、D组均在20%乌拉坦腹腔注射麻醉起效后,行气管切开,插入气管导管(用14~16号静脉穿刺套管针外套管自制)接小动物呼吸机行机械通气,通气时间分别为1、2和4 h,机械通气参数参考Karzai等[6]的相关研究统一设置为:潮气量(VT):40 mL/kg,呼吸频率(RR):40次/min,吸/呼比(I∶E)为 1∶1,PEEP为0,吸入气体为室内空气,潘库溴胺肌注维持肌肉松弛。股静脉和颈内动脉穿刺置管,监测生命体征及补液等。机械通气达到预定时间后,放血处死大鼠。小心分离左右两侧肺组织,左肺行肺灌洗,右侧上叶肺组织石蜡包埋,其余3叶右肺组织直接放入液氮中保存待检。回收的肺灌洗液(BALF)一部分行白细胞计数,另一部分立即离心10 min(2 500 r/min),上清液于-70℃冰箱保存待检。
1.3 主要测定指标及方法
1.3.1 肺组织病理学观察 将上述石蜡包埋的肺组织标本切片行苏木精-伊红(hematoxylin-eosin,HE)染色后,光学显微镜下观察肺组织结构、肺内炎性细胞浸润及分类等特征改变。按以下4项指标行急性肺损伤(ALI)评分[7],A:肺毛细血管充血;B:肺内出血;C:中性粒细胞在血管壁肺间隙集聚或浸润;D:肺泡壁增厚或透明膜形成。每一项按5分制来判断:0=最轻微损害;1=轻微损害;2=中度损害;3=重度损害;4=最严重损害。
1.3.2 肺湿干重(W/D)比值测定及中性粒细胞计数 取动物一叶右肺组织,称湿重后置于烤箱中,70℃烘烤至恒重后称干重,计算肺W/D比值;肺灌洗方法:2 mL生理盐水缓慢注入左肺,片刻后回抽,反复3次,然后回收BALF行白细胞计数。
1.3.3 总蛋白和髓过氧化物酶(MPO)活性测定
BALF中总蛋白的测定采用考马斯亮蓝染色法,采用UV-2000型分光光度计测量吸光度;肺组织MPO活性测定采用MPO试剂盒:准确称取液氮保存的大鼠肺组织0.1 g,按重量体积比1∶19加匀浆缓冲液制备成5%组织匀浆,按照试剂盒说明书步骤,分光光度计法测定吸光度,计算出MPO活性。
1.3.4 RT-PCR检测肺组织中血管紧张素原(AGT)、ACE及AT1mRNA的表达水平 准确称取100 mg肺组织,Trizol一步法提取RNA;所得的RNA产物溶于50 μ L DEPC-H2O中,测量样品在260和280 nm波长下的吸光度值,确定样品RNA的质量和纯度;抽取1 μ g总RNA,严格按逆转录试剂盒所提供的方法合成 cDNA;PCR反应体系包括 :cDNA 4 μ L,Buffer 5 μ L,dNTP 1 μ L,引物的正义链和反义链各 1 μ L,MgCl24 μ L,Taq 酶 1 μ L,加水至50 μ L。内参照管家基因磷酸甘油醛脱氢酶(GAPDH)引物序列为:5′-TGAAGGTCGGTGTCAACGGAT T TGGC-3′和 5′-CATGTAGGCCATGAGGTCCACCAC-3′,目的基因片段长度为 983 bp;AGT 特异引物 为:5′-CCTCGCTCTCTGGACT TATC-3′和 5′-CAGACACTGAGGTGCT-GTTG-3′,目的基因片段长度为226 bp;ACE特异引物为:5′-GCT TCCCCAACAAGACTGCCA-3′和5′-CCACATGTCTCCCAGCAGATG-3′,目的基因片段长度为 380 bp;AT1特异性引物为:5′-GTAGCCAAAGTCACCTGCAT-3′和 5′-TCAGGCAATTGT TAAAATAAGCTAT-3′,目的 基 因片段长度为463 bp。PCR扩增条件:95℃预变性。变性:94℃,30 s;退火:55℃,30 s(GAPDH)/60℃,30 s(AGT)/57℃,30 s(AT1);延伸:72℃,30 s(AT1)/72℃,45 s(其他)。72℃延伸,4℃终止反应。PCR产物用1.5%琼脂糖凝胶电泳,凝胶成像系统采集图像,图像分析软件(Imagemaster)分析目的基因片段的吸光度值,并计算其与GAPDH值的比值 ,以±s表示。
1.3.5 Western blot法检测ACE蛋白的含量 少量大鼠右肺组织与液氮混合后碾碎,提取核蛋白;考马斯亮蓝法定量。SDS-聚丙烯酰胺凝胶恒压电泳;蛋白转膜后一抗孵育,4℃过夜;室温下二抗摇床杂交1 h;ECL化学发光法检测阳性信号。采用Bio-Rad Gel Doc1000成像系统采集X线片上的试验结果图像,用图像分析软件(Image master)分析图像条带,并计算其积分吸光度。
1.3.6 酶联免疫吸附试验(ELISA)法测定肺组织中ANGⅡ的水平 取肺组织匀浆,严格按照试剂盒说明操作,在酶标仪上测出标准品吸光度并绘制出标准曲线,然后根据测得的样品吸光度在标准曲线上读出样品的浓度(pg/mL)。
1.3.7 免疫组化法检测肺组织AT1的表达 采用链霉亲合素-生物素化过氧化酶复合物法(SABC法),按照北京中山公司SABC试剂盒操作步骤进行;在400倍光镜下每张切片随机观察10个视野,细胞质和细胞核呈棕黄色为阳性;计数每100个细胞中阳性细胞的数量。
1.4 统计学处理
2 结果
2.1 病理学改变肺损伤评分(ALI评分)
A组肺组织无明显病理改变,肺泡间隔正常,偶见少量的炎性细胞及巨噬细胞,肺泡腔无渗出物;B组肺组织病理改变明显,肺泡间隔增厚,肺泡腔内可见较多的炎性细胞浸润;C组肺组织病理改变较B组加重,肺泡间隔明显增厚,肺泡腔浸润的炎性细胞明显增多,部分肺泡萎陷或破裂;D组肺组织病理改变又较C组加重,肺泡及肺间质水肿,部分肺泡隔断裂、肺泡破裂、相互融合,大量肺泡萎陷且程度不均一。和A组相比,B、C、D组ALI评分均显著增高(均P<0.01);和B组比较,C、D两组 ALI评分显著增高(均P<0.05);和C组比较,D组ALI评分也显著增高(P<0.05)。见表1。
2.2 肺W/D及肺组织(BALF)中MPO、总蛋白浓度、中性粒细胞计数值
和A 组相比,B、C、D 组W/D、MPO、总蛋白浓度、中性粒细胞计数值显著升高(均P<0.01);和B组相比,C、D组W/D、MPO、总蛋白浓度、中性粒细胞计数值亦显著升高(均P<0.05);和C组相比,D组W/D、MPO、总蛋白浓度、中性粒细胞计数值显著升高(均P<0.05)。见表1。
表1 各组肺组织ALI评分、湿干重比值、M PO活性,BALF中总蛋白、白细胞计数的比较(±s,n=6)Table 1 Comparison of ALI scores,wet/dry ratios,MPO activity,total protein and neutrophil counts(±s,n=6)
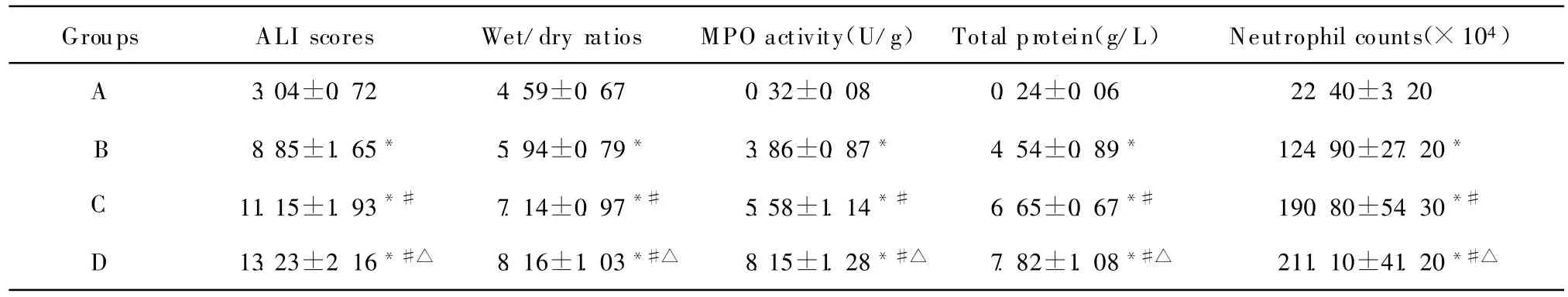
表1 各组肺组织ALI评分、湿干重比值、M PO活性,BALF中总蛋白、白细胞计数的比较(±s,n=6)Table 1 Comparison of ALI scores,wet/dry ratios,MPO activity,total protein and neutrophil counts(±s,n=6)
*P<0.01 vs g roup A;#P<0.05 vs g roup B;△P<0.05 vs group C
Groups ALI scores Wet/dry ratios MPO activity(U/g) Total protein(g/L) Neutrophil counts(×104)A 3.04±0.72 4.59±0.67 0.32±0.08 0.24±0.06 22.40±3.20 8.85±1.65* 5.94±0.79* 3.86±0.87* 4.54±0.89* 124.90±27.20*C 11.15±1.93*# 7.14±0.97*# 5.58±1.14*# 6.65±0.67*# 190.80±54.30*#B D 13.23±2.16*#△ 8.16±1.03*#△ 8.15±1.28*#△ 7.82±1.08*#△ 211.10±41.20*#△
2.3 RT-PCR检测肺组织AGT、ACE、AT1mRNA的表达水平
和A 组比较,B、C、D 组 AGT 、ACE、AT1mRNA的表达水平显著增高(均P<0.01);和B组比较,C、D两组AGT、ACE、AT1mRNA的表达水平显著增高(均P<0.05);和C组比较,D组AGT、ACE、AT1mRNA的表达水平显著增高(均 P<0.05)。见图1、表2。

图1 RT-PCR检测各组肺组织AGT、ACE、AT1mRNA的表达水平Fig.1 The expressions of AGT,ACE,A T1mRNA in lung tissues of each group detectd by RT-PCR
2.4 Western blot检测肺组织ACE蛋白的含量
和A组比较,B、C、D组肺组织ACE蛋白的含量显著增高(均P<0.01),和B组比较,C、D两组ACE蛋白的含量显著增高(均P<0.05);和C组比较,D组ACE蛋白的含量亦显著增高(P<0.05)。见图2、表2。

图2 Western blot检测各组肺组织ACE蛋白的含量Fig.2 The expressions of ACE protein in lung tissues of each g roup detected by Western blot
2.5 ELISA检测大鼠肺组织ANGⅡ的含量
和A组比较,B、C、D组肺组织ANG Ⅱ含量显著增多(均P<0.01);和B组比较,C、D组ANG Ⅱ水平显著升高(均 P<0.05);和C组比较,D组ANG Ⅱ水平亦显著增高(P<0.05)。见表2。
2.6 免疫组化法检测肺组织AT1的表达
A组肺组织肺上皮细胞AT1仅有微弱的表达,而B、C、D组肺组织肺上皮细胞AT1表达显著增多(均 P<0.05);和B组相比,C、D组 AT1表达水平显著升高(均P<0.01);C和D组AT1的表达水平差异无统计学意义。见图3、表2。
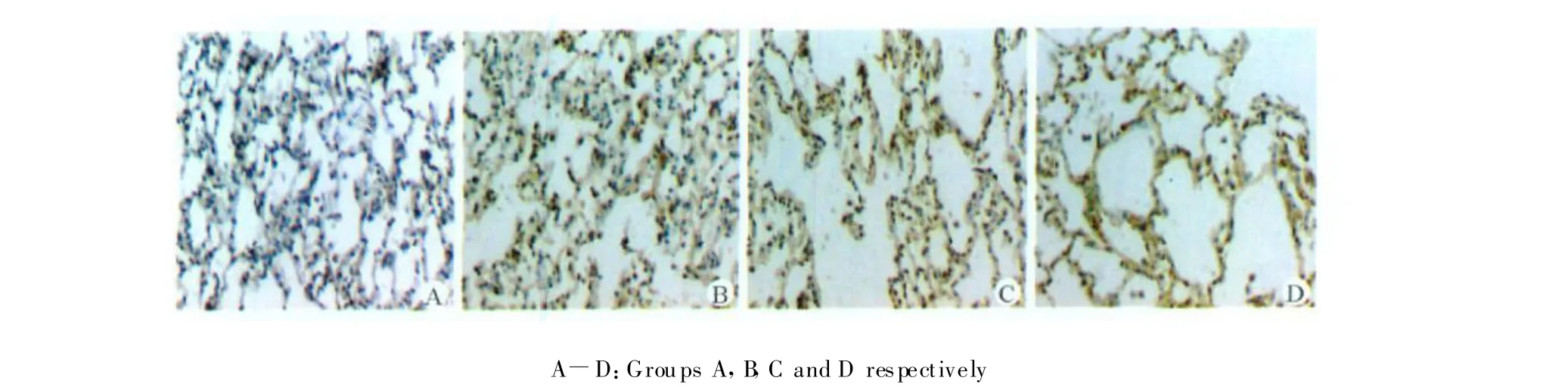
图3 免疫组化法检测各组肺组织AT1的表达(SABC染色,×200)Fig.3 T he ex pressions of AT1protein in lung tissues of each group detected by immunohistochemical techniques(SABC staining,×200)
表2 各组肺组织AGT、ACE、AT1mRNA的表达水平、ACE蛋白、ANG Ⅱ的含量及AT1阳性细胞计数的比较(±s,n=6)Table 2 Comparison of the expression of AGT,ACE,AT1mRNA,ACE protein,ANG Ⅱ contents and AT1 positive cell counts(±s,n=6)

表2 各组肺组织AGT、ACE、AT1mRNA的表达水平、ACE蛋白、ANG Ⅱ的含量及AT1阳性细胞计数的比较(±s,n=6)Table 2 Comparison of the expression of AGT,ACE,AT1mRNA,ACE protein,ANG Ⅱ contents and AT1 positive cell counts(±s,n=6)
*P<0.01 vs g roup A;#P<0.05 vs g roup B;△P<0.05 vs group C
Groups AG T mRNA ACE mRNA A T1mRNA ACE protein ANGⅡ(pg/mL) AT1positive cell A 0.15±0.03 0.16±0.04 0.18±0.05 22.03±4.15 115.63±17.25 12.56±3.57 B 0.29±0.06* 0.31±0.06* 0.33±0.07* 49.31±3.56* 223.84±33.51* 30.72±5.36*C 0.45±0.11*# 0.50±0.12*# 0.58±0.13*# 112.48±5.60*# 328.17±49.26*# 56.22±6.49*#D 0.65±0.16*#△ 0.79±0.17*#△ 0.81±0.18*#△ 169.75±8.17*#△ 416.32±56.53*#△ 79.61±8.13*#
3 讨论
VILI是肺功能严重受损的患者行机械通气支持治疗时的常见并发症,其致病机制非常复杂,包含急性炎症反应、氧化应激反应、细胞凋亡等多方面的因素,与其他致病因素(如:内毒素)所致肺损伤的致病机制略有不同[8-9]。
在本研究中,对照组肺组织病理切片无明显病理改变,肺组织ALI评分处于较低水平。而B、C、D组肺组织病理切片出现明显的急性炎性损伤性改变,具体表现为肺间隔明显增厚、炎性细胞的浸润以及肺泡腔中出现渗出物等,且其程度(ALI评分)随着机械通气时间的延长而显著加重。W/D比值和BALF中总蛋白可反映肺水肿的程度;MPO是中性粒细胞释放的一种过氧化物酶类,和中性粒细胞的数目存在极显著的相关性,可反映肺组织中性粒细胞浸润的水平。我们的实验结果发现,和对照组相比,B、C、D 组肺组织 W/D、BALF中MPO 活性、总蛋白浓度、中性粒细胞计数值显著增高。并随着通气时间的延长而显著增加,这些结果和Karzai等[6]的研究一致,说明我们成功地复制了大鼠VILI模型。且大鼠肺损伤程度和机械通气的时间密切相关,大鼠机械通气4 h时VILI最严重,我们的预实验也发现,如果机械通气时间超过4 h,大鼠死亡率会显著增高。
近些年来,RAS在炎症反应、急性肺损伤中的作用逐渐受到关注。RAS不只存在于心血管系统,研究表明,在人和大鼠肺组织中,也存在局部RAS,并对肺组织细胞的生长、凋亡起着重要的调节作用,其中尤以ANGⅡ及AT1较为关键。ANG Ⅱ最初是作为一种具有血管收缩作用的多肽被发现,近些年来发现ANGⅡ及其受体广泛参与了机体的炎症反应、氧化应激、细胞凋亡等多方面的作用,具有较为广泛的生物学效应。研究证明,ANGⅡ通过激活AT1,能诱导血管内皮细胞、平滑肌细胞内多种和炎症密切相关的信号转导通路(如p38MAPK、JNKs等)的激活[10-11]。细胞和动物实验证明,ANGⅡ通过激活AT1可使多种细胞间黏附分子、炎性细胞因子、趋化因子的表达显著增高,提示ANG Ⅱ可能在炎症反应的机制中起着重要的作用[12-13]。正因为RAS在急性炎症反应、氧化应激、细胞凋亡中起着多方面的作用,RAS在急性肺损伤致病机制中的作用才逐渐受到重视。
在本研究结果中:和对照组相比,B、C、D组肺组织AGT、ACE、AT1mRNA 的表达水平、ACE及ANG Ⅱ的含量显著增高(均P<0.01),且上述各指标可随着通气时间的延长而显著增加。结果说明大鼠大潮气量机械通气在产生了明显VILI的同时,也显著激活了RAS;我们同时发现:对照组肺组织肺上皮细胞AT1仅有较微弱的表达,而B、C、D组肺组织肺上皮细胞 AT1表达显著增多(均 P<0.05);和B组相比,C、D组AT1表达水平显著升高(均P<0.05);可能是VILI激活了RAS后诱导大鼠肺组织AGT、ACE、ANG Ⅱ及AT1的表达显著增加,这可能是VILI的致病机制之一,但其具体作用机制还需更进一步研究。
[1] Wang R,Zagariya A,Ibarra-Sunga O,et al.AngiotensinⅡinduces apoptosis in human and rat alveolar epithelial cells[J].Am J Physiol,1999,276(5):885-889.
[2] Imai Y,Kuba K,Rao S,et al.Angiotensin-converting enzyme 2 protects from severe acute lung failure[J].Nature,2005,436(7):112-116.
[3] Raiden S,Nahmod K,Nahmod V,et al.Nonpeptide antagonists of AT1receptor for angiotensinⅡdelay the onset of acute respiratory distress syndrome[J].J Pharmacol Exp T-her,2002,303(1):45-51.
[4] Lukkarinen H,Laine J,Lehtonen J,et al.AngiotensinⅡreceptor blockade inhibits pneumocy te apoptosis in experimental meconium aspiration[J].Pediatric Res,2004,55(2):326-333.
[5] Marshall R P,Webb S,Bellingan G J,et al.Angiotensin converting enzyme insertion/deletion polymorphism is associated with susceptibility and outcome in acute respiratory distress sy ndrome[J].Am J Respir Crit Care Med,2002,166(5):646-650.
[6] Karzai W,Cui X Z,Heinicke N,et al.Neutrophil stimulation with granulocyte colony-stimulating factor worsens ventilator-induced lung injury and mortality in rats[J].Anesthesiology,2005,103(4):996-1005.
[7] Murray J F,Matthay M A,Luce J M,et al.An expanded definition of the adult respiratory distress syndrome[J].Am Rev Respir Dis,1988,138(3):720-723.
[8] Wösten-van Asperen R M,Lutter R,Haitsma J J,et al.ACE mediates ventilator-induced lung injury in rats via angiotensinⅡbut not bradykinin[J].Eur Respir J,2008,31(2):363-371.
[9] Wang Y L,Yao S L,Xiong P.Expression changes of early response genes in lung due to high volume ventilation[J].J Huazhong Univ Sci T echnol[Med Sci],2005,25(3):339-342.[10] Chen C M,Chou H C,Wang L F,et al.Captopril decreases plasminogen activator inhibitor-1 in rats with ventilator-induced lung injury[J].Crit Care Med,2008,36(6):1880-1885.[11] T ouy z R M,He G,Elmabrouk M,et al.Differential activation of ERK1/2 and p38M AP kinase by angiotensinⅡtypeⅠreceptor in vascular smooth muscle cells from WKY and SHR[J].J Hypertens,2001,19(3 Pt 2):553-559.
[12] Alvarez A,Cerdá-Nicolás M,Naim Abu Nabah Y,et al.Direct evidence of leukocyte adhesion in arterioles by angiotensinⅡ[J].Blood,2004,104(2):402-408.
[13] Ito T,Ikeda U,Yamamoto K,et al.Regulation of interleukin-8 expression by HMG-CoA reductase inhibitors in human vascular smooth muscle cells[J].Atherosclerosis,2002,165(1):51-55.
猜你喜欢
中国病理生理杂志(2022年7期)2022-08-05
中国民间疗法(2021年14期)2021-08-30
现代临床医学(2021年4期)2021-07-31
临床误诊误治(2021年7期)2021-07-26
国际呼吸杂志(2019年8期)2019-04-29
国际呼吸杂志(2019年8期)2019-04-29
老年医学与保健(2017年6期)2017-02-06
医学研究杂志(2015年3期)2015-06-10
中国当代医药(2015年21期)2015-03-01
中国实用医药(2014年31期)2014-11-12
