
α-Actinin影响丙型肝炎病毒非结构蛋白与脂筏的关联及复制
2010-01-26 06:48宋武慧潘婷婷袁正宏
微生物与感染 2010年4期
宋武慧,潘婷婷,袁正宏
复旦大学上海医学院教育部/卫生部医学分子病毒学重点实验室, 上海200032
丙型肝炎病毒(hepatitis C virus,HCV)感染人体后可在细胞内持续复制,导致慢性肝炎、肝硬化, 最终可能发展成肝细胞肝癌而引起患者死亡[1]。目前认为,HCV编码的多聚蛋白可被切割成病毒的结构蛋白和非结构蛋白(nonstructural protein,NS),其中非结构蛋白在病毒复制中起关键作用[2]。首先HCV NS4B可引起细胞内质网(endoplasmic reticulum,ER) 膜结构变化,形成可耐受核酸酶及蛋白酶的泡膜结构;然后HCV非结构蛋白与宿主蛋白相互作用在此结构上形成复制复合体(replication complex,RC),为HCV复制提供场所[3]。有研究报道,HCV复制复合体中非结构蛋白可与脂筏标志分子共定位于去污剂耐受膜(detergent-resistant membrane,DRM),表明脂筏与HCV复制密切相关[4]。本实验室曾利用蛋白质组学结合生物学分析,分离鉴定含有HCV复制复合体的脂筏成分,发现复制复合体对脂筏删除试剂敏感,该试剂可使NS5A与复制复合体的重要成分VAP-33共定位消失,证实HCV复制复合体与脂筏相关,且NS5A为HCV 复制复合体完整性的重要标志分子[5-7]。
α-actinin广泛存在于肌型和非肌型细胞中,可结合并交联F-肌动蛋白(actin),其中肌型α-actinin可参与肌肉的收缩调节;非肌型α-actinin可参与集结肌动蛋白微丝,形成张力纤维(stress fibre),并将其锚定于细胞膜,是细胞微丝骨架与细胞膜相互作用的重要桥梁,在维持细胞正常形态中起重要作用[8]。已有研究表明,α-actinin第12位为酪氨酸磷酸化位点,当加入局部黏着斑激酶(focal adhesion kinase,FAK)使第12位磷酸化后,原与肌动蛋白紧密交联的α-actinin随即解离。磷脂酰肌醇-3-激酶(phosphoinositide 3-kinase,PI3K)和钙离子可结合α-actinin,使其与肌动蛋白的结合力下降,导致细胞骨架蛋白流动,有利于细胞运动和迁移[9]。此外α-actinin还可与细胞膜黏附分子、细胞膜受体等相互作用,参与细胞黏附、转移性转化、膜受体表达等多种生理过程[10]。
本研究室前期研究发现,α-actinin参与调控HCV亚基因组RNA合成[11],但其参与病毒复制的具体机制尚不清楚。为深入研究α-actinin参与HCV复制的分子机制,本课题分别在复制子系统和感染性病毒颗粒HCVcc系统中证实α-actinin参与HCV复制,随后的膜漂浮实验显示α-actinin可与HCV NS5A共定位于脂筏。利用小干扰RNA(small interfering RNA,siRNA)技术抑制内源性α-actinin表达,NS5A从脂筏脱落。免疫荧光结果显示,NS5A与内质网标志分子calnexin核周共定位消失。以上研究初步探讨了α-actinin参与HCV复制的机制,为深入阐明HCV复制机制打下了基础。
1 材料和方法
1.1 材料
实验材料包括Huh7.5细胞(美国Rockefeller大学Charles Rice教授惠赠),复制子细胞(本室构建保存);DMEM细胞培养基、胎牛血清(Gibco公司),DMSO(Merck公司),青霉素(1×105u/L)、链霉素(0.1 g/L) (Gibco公司),细胞培养瓶、培养皿、培养板(NUNC公司) ;限制性内切酶(NEB公司、MBI公司), TRIzol、TRIzol LS、DEPC、Opti-MEM(Invitrogen公司),Tris饱和酚、氯仿(上海申能博彩公司),ExTaqHS酶(TaKaRa公司) ,SuperScriptⅡ反转录酶(Invitrogen公司) ,体外转录试剂盒 (Promega公司);转染试剂:FuGene 6(Roche公司),RNAiMAX(Invitrogen公司);anti-actinin抗体(Santa Cruz公司),anti-β-tubulin抗体、anti-actin抗体、anti-HA抗体(Sigma公司),anti-NS3抗体、anti-NS5A(1b) 抗体(ViroGen公司),anti-NS5A 9E10(2a)抗体(Charles Rice教授惠赠),anti-myc抗体(Santa Cruz公司),anti-calnexin抗体、小鼠二抗、大鼠二抗 ( Santa Cruz公司),荧光二抗cy3、Alexa 488(Molecular Probes公司),硝酸纤维素膜(孔径0.22 μm,Schlercher & Schuell BioScience公司);宿主菌Top10F(Invitrogen公司) ,质粒抽提纯化试剂盒(Qiagen公司、Macherey-Negal公司) ,ECL发光(PerkinElmer公司)。
1.2 方法
1.2.1细胞培养Huh7.5和JFH1感染的 Huh7.5细胞培养于含10%胎牛血清、1×105u/L青霉素、0.1 g/L 链霉素的DMEM培养基, 置5% CO2、37 ℃细胞培养箱内培养,2~3 d 传代1次。复制子细胞培养液中再加0.5 μg/ml 的杀稻瘟菌素(blasticidin)。
1.2.2质粒构建以肝文库cDNA为模板,聚合酶链反应(polymerase chain reaction,PCR)扩增α-actinin片段,经EcoRⅠ和XhoⅠ双酶切后分别插入2种载体后形成质粒pCMV-HA-α-actinin和pcDNA3.1-α-actinin-myc。质粒均经测序验证。
1.2.3转染siRNA将复制子细胞接种于24孔板,用于蛋白免疫印迹和定量PCR实验;将复制子细胞接种于直径10 cm的培养皿,用于膜漂浮和免 疫荧光实验。无相关RNA作为阴性对照。siRNA 序列(HSS100130)共3种,分别为5′-UUGCAUGGCAUGCAUGGUGUUCUCG-3′;5′-CGAGAACACCAUGCAUGCCAUGCAA-3′;5′-AAUAUCAUAACCCAAGCUGAUGAGG-3′。siRNA转染采用RNAiMAX。具体方法参照说明书,转染6 h后换成完全DMEM继续培养。
1.2.4JFH1感染细胞的建立pJFH1质粒由日本Wakita T教授惠赠。质粒模板线性化后,体外转录JFH1 RNA并电转染入Huh7.5细胞,定期收集病毒并检测病毒滴度。具体方法参照文献[12]。
1.2.5JFH1感染实验将 Huh7.5细胞接种于12孔板,当细胞融合70%~80%时,以FuGene 6转染pCMV-HA-α-actinin,DNA总量1 μg,以转染pCMV-HA和未转染细胞为阴性对照。转染24 h 后将细胞转移到6孔板,12 h后感染HCV(感染复数0.1)。感染后12 h用磷酸缓冲液(phosphate-buffered saline,PBS) 洗3 次,更换新鲜培养液,48 h后收集细胞及培养液。
1.2.6病毒滴度测定将Huh7.5细胞接种于96孔板,过夜贴壁后加入病毒浓缩液;12 h后更换新鲜培养液,48 h后收集细胞;免疫荧光标记NS5A并计数阳性细胞。为保证计数准确,由2人分别各数2次,取平均值,计算病灶形成单位(focus-forming unit,FFU)。具体方法参照文献[12]。
1.2.7蛋白免疫印迹法蛋白样品经10%十二烷基磺酸钠-聚丙烯酰胺凝胶电泳(sodium dodecyl sulfate polyacrylamide gel electrophoresis,SDS-PAGE)分离,电转印至硝酸纤维素膜,5%牛奶(PBS配制)封闭后,分别加anti-NS3抗体、anti-NS5A 抗体、anti-actin抗体、anti-β-tubulin抗体、anti-actinin抗体、anti-myc抗体和anti-HA抗体,置4 ℃ 过夜。用PBST(含0.05% Tween-20的PBS) 洗膜3次,每次10 min,加入相应辣根过氧化物酶(horseradish peroxidase,HRP) 标记的二抗,置室温2 h,PBST 洗膜3 次,ECL 发光。
1.2.8荧光定量PCR用TRIzol 裂解 JFH1感染细胞,用TRIzol LS裂解细胞培养液。分别加入对应量氯仿,震荡混匀,12 800g离心15 min。取上层水相,加适量异丙醇沉淀RNA,12 800g离心15 min。300 μl 70%乙醇洗涤沉淀,12 800g离心15 min。弃液体,沉淀干燥后加SuperScript Ⅱ体系,快速去除基因组DNA 并进行反转录。取2 μl 反转录cDNA与相应引物(sense: 5′-CCCTGTGA- GGAACTATCTGTCTTCACGC-3′; antisense: 5′-TCCAGAGCATCTGGCACGTA/GGTACTCG-3′),加至实时荧光定量PCR 反应液中(总体积18 μl)。样品95 ℃变性3 min ;先按95 ℃5 s、55 ℃10 s、72 ℃20 s 进行3个循环,接着按95 ℃15 s、60 ℃30 s、79 ℃8 s进行36个循环。在每个循环结束前系统自动检测荧光产物的量,所有循环结束后绘制熔解曲线,判断扩增产物的特异性。用ABI 7500 software进行数据处理,对两样本均数t检验,P<0.05为有显著统计学差异。每组数据重复3次。
1.2.9膜漂浮实验首先将1.5×107个复制子细胞用PBS 洗1次,在冰上刮取细胞,4 ℃,900g离心5 min。收集细胞,沉淀物用0.95 ml 预冷低渗缓冲液 (10 mmol/L Tris-HCl,pH 7.8;10 mmol/L NaCl;EDTA-free protease inhibitor cocktail)悬浮,冰上放置15 min使细胞溶胀,用玻璃匀浆器抽吸20次。细胞裂解液4 ℃,900g离心5 min。弃沉淀物,上清液加入50 μl 20%的NP-40,使其终浓度为1%,冰上放置30 min。总体积1 ml的悬液与3 ml 72% (W/W)蔗糖溶液(50 mmol/L Tris-HCl,pH7.5;25 mmol/L KCl;5 mmol/L MgCl2)混匀,其上逐步添加4 ml 55% (W/W)、1.5 ml 10% (W/W)蔗糖溶液。离心管多余部分用液状石蜡补平,4 ℃,24 000g离心16 h。从蔗糖梯度顶端依次取1 ml,共9组,每组加4 ml 冰冷100%甲醇,混匀,10 000g离心10 min。沉淀物加50 μl 2×SDS Loading Buffer,煮沸10 min;取15 ml 进行10% SDS-PAGE分离,然后用针对α-actinin、NS5A和myc的抗体进行蛋白免疫印迹实验,检测蛋白条带。
1.2.10免疫荧光实验实验玻片的处理:无水乙醇浸泡盖玻片,用10 μg/ml 的poly-D-lysine 37 ℃处理过夜。细胞收集后,PBS洗1次,加入3.5%聚合甲醛,室温固定15 min;吸去固定液,加30 mmol/L甘氨酸(PBS配制)终止反应5 min;PBS洗3次,加PBS+0.1% Triton X-100室温穿透5 min;PBS洗2次,加3% PBS-BSA封闭1 h;加一抗NS5A和calnexin(PBS-BSA稀释),4 ℃过夜;PBS洗3次,每次5 min,加荧光二抗cy3和Alexa 488室温孵育1 h;PBS洗3次,每次10 min,用4’,6-二脒基-2-苯基吲哚(4’,6-diamidino-2-phenylindole,DAPI)(1∶5 000,用PBS稀释)染色,Mowiol封片,共聚焦观察(Leica,TCS-NT,Heidelberg,Germany)。
2 结果
2.1 抑制α-actinin表达导致HCV非结构蛋白减少
为抑制内源性α-actinin表达,首先对siRNA-actinin序列进行选择。在HCV复制子细胞中分别转入3组siRNA-actinin,与无相关对照相比,3组α-actinin表达皆受抑制。其中转染siRNA-actinin 2(5′-CGAGAACACCAUGCAUGCCAUGCAA-3′)后,α-actinin表达显著减少,因此以该序列作为siRNA-actinin(图1A)。
在HCV复制子细胞中转入siRNA-actinin,无相关对照siRNA为阴性对照,48 h后收集细胞,用蛋白免疫印迹法检测α-actinin及HCV非结构蛋白的表达。结果显示,与对照组相比,抑制细胞内α-actinin表达导致NS5A和NS3表达均减少(图1B)。HCV RNA水平亦受抑制,与本室已报道的结果一致[11]。
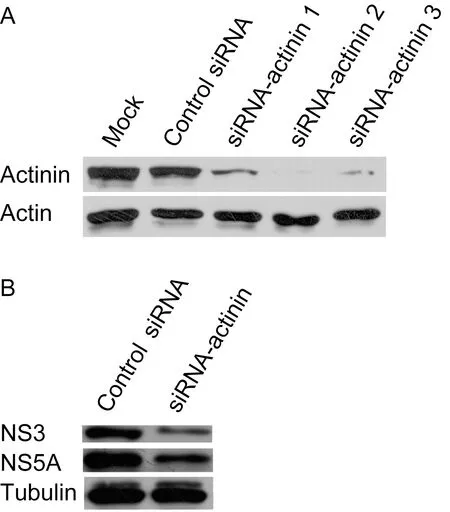
HCV replicon cells were transfected with control siRNA or siRNA-actinin. At 48 h post-transfection, the cells were harvested. A: Immunoblotting analysis of protein expression level with antibodies against actinin. Actin was detected as a loading control. B: Immunoblotting analysis of protein expression level with antibodies against NS3 and NS5A. Tubulin was detected as a loading control.
图1抑制α-actinin表达导致HCV非结构蛋白减少
Fig.1Knockdownofα-actinininhibitedHCVnonstructuralproteinexpressioninrepliconcells
2.2 过表达α-actinin可上调JFH1中HCV RNA水平及其非结构蛋白表达
为验证α-actinin是否仅参与HCV1b亚型的调控,我们进一步在Huh7.5细胞中转染 pCMV-HA-α-actinin,以转染pCMV-HA和未转染细胞为阴性对照。转染24 h 后将细胞转移到6孔板,12 h后感染HCV2a亚型病毒(感染复数0.1),感染后12 h更换新鲜培养液。48 h后收集细胞和细胞培养液,蛋白免疫印迹和定量PCR法分别检测α-actinin质粒及HCV非结构蛋白的表达和HCV RNA水平;病毒滴度测定方法检测细胞培养液中病毒颗粒感染性。结果显示,Huh7.5细胞转染pCMV-HA-α-actinin后,蛋白表达增加(图2A)。与对照组相比,当α-actinin表达增加时,NS5A表达随之增加,细胞内和细胞培养液中HCV RNA 水平亦同时增加,约为阴性对照的2倍(P<0.05,图2B、C),培养液中HCV滴度(ffu/ml)也显著增高,约为阴性对照的4倍(P<0.05,图2D)。
2.3 复制子细胞中α-actinin亚细胞定位于脂筏
已知HCV非结构蛋白与宿主蛋白相互作用可在脂筏上组装成复制复合体[4]。为探讨α-actinin在HCV复制子细胞中的定位,我们分别瞬时转染全长pcDNA3.1-α-actinin-myc和pcDNA3.1-myc,48 h后收集细胞,用不连续蔗糖密度梯度法分离细胞中的脂筏成分。结果显示,α-actinin质粒可在细胞内表达,且与NS5A位于相同膜组分,共定位于脂筏(图3)。免疫荧光结果也显示,α-actinin与内质网标志分子calnexin核周共定位(结果未展示)。
2.4 抑制α-actinin表达影响HCV非结构蛋白的膜定位
鉴于α-actinin具有骨架蛋白特性且定位于脂筏,我们推测其可能通过影响HCV非结构蛋白的亚细胞定位来调控HCV复制。为证实该假设,在HCV复制子细胞中转入siRNA-actinin,无相关siRNA为阴性对照,48 h后收集细胞,分别用膜漂浮和免疫荧光实验检测HCV复制复合体重要成分之一——NS5A的细胞定位。结果显示,阴性对照组中内源性α-actinin与NS5A共定位于去污剂耐受膜组分,与图3一致;而转染siRNA-actinin组中,α-actinin细胞内表达受抑制,同时NS5A蛋白曝光8 min,结果显示其脂筏定位改变,原本定位于脂筏的NS5A 约50%从去污剂耐受膜组分脱落至去污剂不耐受膜组分(图4A);NS5A典型核周分布消失,未发现与calnexin共定位(图4B)。结果提示,α-actinin的减少可影响复制复合体组成蛋白NS5A的膜定位。
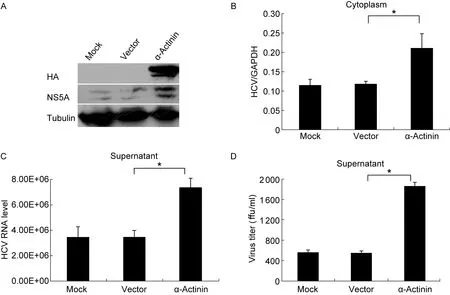
Huh7.5 cells were untransfected (Mock) and transfected with pCMV-HA (Vector) or pCMV-HA-α-actinin. At 24 h post-transfection, the cells were trypsonized and transferred to 6-well plates. The virus suspension was added and the medium was switched to fresh complete DMEM at 12 h post-infection. The cells and supernatant were harvested at 48 h post-infection. A: Immunoblotting analysis of protein expression level with antibodies against NS5A and HA. Tubulin was detected as a loading control. B, C: Cells and supernatant were treated with TRIzol and TRIzol LS reagent, respectively. The real-time PCR signals were analyzed using ABI 7500 software.*P<0.05. D: Titration of infectious HCV for NS5A expression was used to quantitate the amount of HCVcc infectivity as ffu/ml.*P<0.05.
图2过表达α-actinin可上调JFH1中HCVRNA水平及其非结构蛋白表达
Fig.2Overexpressionofα-actininupregulatedHCVRNAlevelandnonstructuralproteinexpression
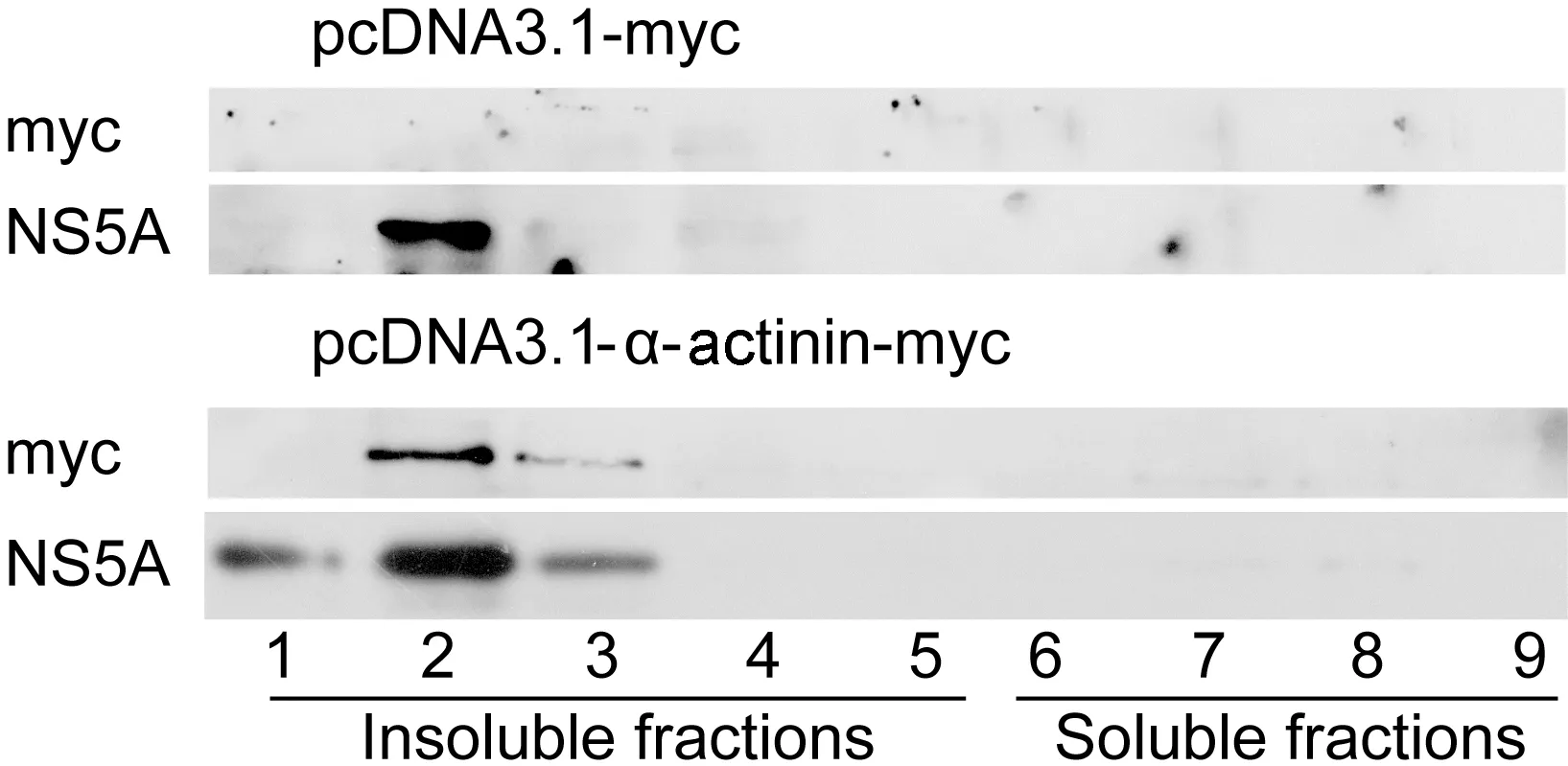
HCV replicon cells were transfected with pcDNA3.1-myc or pcDNA3.1-α-actinin-myc. At 48 h post-transfection, the cells were collected for membrane flotation analysis. Immunoblotting analysis of protein expression level with antibodies against myc and NS5A.
图3复制子细胞中α-actinin亚细胞定位于脂筏
Fig.3α-actininwaslocatedinlipidraftinrepliconcells
3 讨论
本研究分别在1b型HCV复制子和2a型JFH1 HCVcc系统中发现,过表达α-actinin可显著增加HCV RNA水平及非结构蛋白表达;抑制α-actinin的表达使HCV非结构蛋白从脂筏脱落、HCV RNA水平下降,提示α-actinin可参与HCV复制,且具有普遍性,不局限于某HCV亚型。
α-actinin可能参与HCV复制复合体在脂筏上的形成。与一般甘油磷脂膜结构不同,脂筏富含胆固醇和鞘磷脂成分,使其可在低温下抵抗非离子去污剂NP-40的作用。脂筏主要功能是参与信号转导和胞内小分子膜运输,一些病毒利用脂筏的特殊性和功能来进行入胞、装配等[13, 14]。本研究发现,HCV复制子中α-actinin定位于脂筏,当其表达受抑制,NS5A 对非离子去污剂敏感而易从脂筏脱落;免疫荧光实验也显示NS5A与calnexin共定位消失。由于α-actinin在细胞微丝骨架与细胞膜结构的相互作用中起桥梁作用,推测其有助于HCV复制复合体锚定于细胞骨架或膜状结构,在亚细胞隔间促进、聚集复制复合体组成蛋白而形成高效复制复合体。此外,有研究表明,破坏肌动蛋白网络(actin network)可导致脂筏标志分子caveolin-2对非离子去污剂敏感,提示脂筏可能受破坏[15]。α-actinin属肌动蛋白交联蛋白家族,可通过肌动蛋白结合结构域(actin-binding domain,ABD)与F-肌动蛋白结合,形成α-actinin交联肌动蛋白网络[16]。因此,α-actinin是否通过其介导形成的肌动蛋白网络影响脂筏形成,参与HCV复制复合体在脂筏膜上的定位和完整性,进而调控HCV复制值得进一步深入研究。
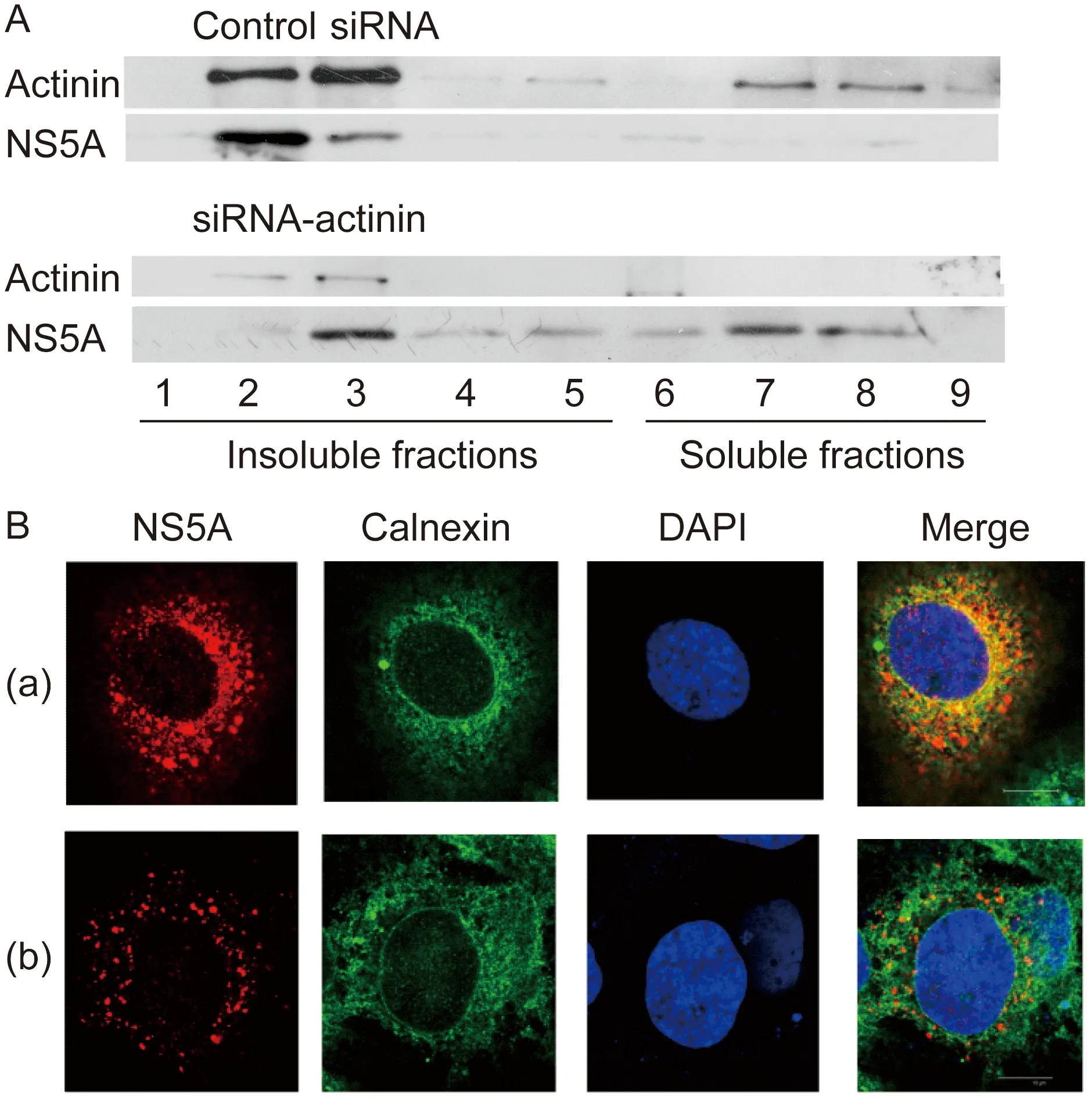
HCV replicon cells were transfected with control siRNA (a) or siRNA-actinin (b). At 48 h post-transfection, the cells were collected for membrane flotation and immunofluorescence analysis. A: Immunoblotting analysis of protein expression level with antibodies against NS5A and actinin. B: The cells were immunostained against NS5A and calnexin, followed by the addition of secondary antibodies conjugated with cy3 (red), Alexa 488 (green), and DAPI (blue). Scale bar, 10 μm.
图4α-actinin表达降低影响HCVNS5A的膜定位
Fig.4Knockdownofα-actininaffectedlocationofHCVNS5Aindetergent-resistantmembraneandendoplasmicreticulum
已有报道认为一些病毒如Andes virus (ANDV)的复制依赖肌动蛋白网络,肌动蛋白多聚化抑制剂细胞松弛素D(cytochalasin D)影响病毒N蛋白的核周定位并下调病毒RNA水平[17]。Bost等发现肌动蛋白多聚化抑制剂可显著抑制HCV RNA的合成;Lai等也发现HCV的NS3和NS5A可结合肌动蛋白,当其完整网络结构被破坏后,将影响NS5A与calnexin共定位,推测HCV非结构蛋白依赖于肌动蛋白网络而在胞质内进行亚细胞器定位和运输[15]。α-actinin与肌动蛋白网络密切相关,是否由其交联形成肌动蛋白网络,通过影响复制复合体组成蛋白的亚细胞定位,从而影响HCV复制复合体的形成,并参与HCV复制,值得进一步研究。
脂滴与HCV装配密切相关。虽然HCV包装和释放过程仍不清楚,但普遍认为HCV NS5A被招募至脂滴结构,通过创造有利于HCV包装的微环境和介导RNA与核衣壳的组装参与装配[18-20]。本研究发现,α-actinin影响NS5A的亚细胞定位,推测NS5A可能通过α-actinin介导的细胞骨架被运输至脂滴结构,参与HCV装配。
综上所述,在前期研究发现α-actinin参与HCV亚基因组复制的基础上,本研究进一步证实α-actinin可通过影响HCV复制复合体中组成蛋白的脂筏定位而参与HCV复制。下一步将对HCV如何利用α-actinin参与其复制的分子机制进行研究。
[1] Beard MR, Helbig KJ. Control of HCV replication: when size does not matter [J]. Hepatology, 2008, 47(3): 1092-1094.
[2] Poenisch M, Bartenschlager R. New insights into structure and replication of the hepatitis C virus and clinical implications [J]. Semin Liver Dis, 2010, 30(4): 333-347.
[3] Brass V, Gosert R, Moradpour D. Investigation of the hepatitis C virus replication complex [J]. Methods Mol Biol, 2009, 510: 195-209.
[4] Yang W, Huang M. Studying HCV RNA synthesis in vitro with replication complexes [J]. Methods Mol Biol, 2009, 510: 177-184.
[5] Yi Z, Fang C, Pan T, Wang J, Yang P, Yuan Z. Subproteomic study of hepatitis C virus replicon reveals Ras-GTPase-activating protein binding protein 1 as potential HCV RC component [J]. Biochem Biophys Res Commun, 2006, 350(1): 174-178.
[6] Fang C, Yi Z, Liu F, Lan S, Wang J, Lu H, Yang P, Yuan Z. Proteome analysis of human liver carcinoma Huh7 cells harboring hepatitis C virus subgenomic replicon [J]. Proteomics, 2006, 6(2): 519-527.
[7] Chang K, Wang T, Luo G. Proteomics study of the hepatitis C virus replication complex [J]. Methods Mol Biol, 2009, 510: 185-193.
[8] Sjoblom B, Salmazo A, Djinovic-Carugo K. Alpha-actinin structure and regulation [J]. Cell Mol Life Sci, 2008, 65(17): 2688-2701.
[9] Esue O, Tseng Y, Wirtz D. Alpha-actinin and filamin cooperatively enhance the stiffness of actin filament networks [J]. PLoS One, 2009, 4(2): e4411.
[10] Low SH, Mukhina S, Srinivas V, Ng CZ, Murata-Hori M. Domain analysis of alpha-actinin reveals new aspects of its association with F-actin during cytokinesis [J]. Exp Cell Res, 2010, 316(12): 1925-1934.
[11] Lan S, Wang H, Jiang H, Mao H, Liu X, Zhang X, Hu Y, Xiang L, Yuan Z. Direct interaction between alpha-actinin and hepatitis C virus NS5B [J]. FEBS Lett, 2003, 554(3): 289-294.
[12] Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. Robust hepatitis C virus infection in vitro [J]. Proc Natl Acad Sci USA, 2005, 102(26): 9294-9299.
[13] Das S, Chakraborty S, Basu A. Critical role of lipid rafts in virus entry and activation of phosphoinositide 3’ kinase/Akt signaling during early stages of Japanese encephalitis virus infection in neural stem/progenitor cells [J]. J Neurochem, 2010, 115(2): 537-549.
[14] Lin S, Wang XM, Nadeau PE, Mergia A. HIV infection upregulates caveolin 1 expression to restrict virus production [J]. J Virol, 2010, 84(18): 9487-9496.
[15] Lai CK, Jeng KS, Machida K, Lai MM. Association of hepatitis C virus replication complexes with microtubules and actin filaments is dependent on the interaction of NS3 and NS5A [J]. J Virol, 2008, 82(17): 8838-8848.
[16] Courson DS, Rock RS. Actin cross-link assembly and disassembly mechanics for alpha-actinin and fascin [J]. J Biol Chem, 2010, 285(34): 26350-26357.
[17] Ramanathan HN, Jonsson CB. New and Old World hantaviruses differentially utilize host cytoskeletal components during their life cycles [J]. Virology, 2008, 374(1): 138-150.
[18] Benga WJA, Krieger SE, Dimitrova M, Zeisel MB, Parnot M, Lupberger J, Hildt E, Luo G, McLauchlan J, Baumert TF, Schuster C. Apolipoprotein E interacts with hepatitis C virus nonstructural protein 5A and determines assembly of infectious particles [J]. Hepatology, 2009, 51(1): 43-53.
[19] Suzuki T, Masaki T, Aizaki H. Involvement of nonstructural protein 5A and lipids on production of hepatitis C virus particles [J]. Uirusu, 2008, 58(2): 199-205.
[20] Hughes M, Griffin S, Harris M. Domain III of NS5A contributes to both RNA replication and assembly of hepatitis C virus particles [J]. J Gen Virol, 2009, 90(Pt6): 1329-1334.
猜你喜欢
中学生物学(2021年8期)2021-11-02
中国畜牧杂志(2020年1期)2020-01-16
中国果业信息(2019年1期)2019-01-05
中国畜牧兽医文摘(2018年6期)2018-07-28
中国病理生理杂志(2018年6期)2018-01-22
中华老年口腔医学杂志(2016年3期)2017-01-15
浙江农业科学(2016年11期)2016-05-04
原子与分子物理学报(2015年1期)2015-11-24
科学启蒙(2015年8期)2015-08-07
中国医科大学学报(2015年10期)2015-03-01
