
结直肠成纤维细胞通过激活ERK信号通路促进结直肠癌细胞的恶性生物学行为
2024-11-11 00:00:00郗雪艳邓婷杜伯雨
南方医科大学学报 2024年10期


摘要:目的 探究人结直肠成纤维细胞(CCD18-Co)条件培养基对结直肠癌(CRC)细胞恶性进展的影响,为CRC治疗提供思路。方法 利用RTCA、克隆形成和创伤愈合实验测定CRC细胞增殖、克隆形成和迁移能力;Western blotting检测CCD18-Co-CM激活的CRC细胞ATK、ERK和STAT3信号通路,同时检测相应信号通路阻断后CRC细胞增殖、克隆形成和迁移能力;肿瘤球形成实验检测CCD18-Co-CM对CRC细胞成球能力的影响;RT-PCR方法检测CRC细胞干性标志物的表达情况。结果 CCD-18Co-CM能够促进CRC细胞的增殖、克隆形成和迁移能力(Plt;0.05)。CCD-18Co-CM能够增强CRC细胞的成球能力及干性标志物的表达(Plt;0.05)。CCD-18Co-CM 能够激活CRC细胞ERK信号通路(Plt;0.05),ERK信号通路抑制剂SCH772984能够降低CRC细胞增殖、克隆形成和迁移能力及成球能力和干性标志物的表达(Plt;0.05)。结论 人正常结直肠成纤维细胞可通过激活ERK通路促进CRC细胞的恶性进展。
关键词:结直肠成纤维细胞;结直肠癌;干性;ERK
结直肠癌(CRC)是一种常见的恶性肿瘤,多发生在直肠-结肠交界处。据2020年统计,全球CRC发病率正以每年2%~4%的速度增长,其患病率和病死率目前位居前3。预计到2030 年,全球CRC患者人数预计将增加60%,发病率和死亡率显著增加[1, 2]。目前,CRC的早期诊断仍具有挑战性,且复发和转移的高发也是影响CRC预后不良的主要原因。肿瘤微环境(TME)是肿瘤细胞赖以生存的土壤和环境,与CRC的发生和发展密切相关[3]。TME 中含量最多的基质细胞是成纤维细胞[4, 5],其主要功能是对组织损伤做出反应并释放生长因子,以促进再生修复,同时维持各种组织结构和功能,但在肿瘤周围和内部高达80%的成纤维细胞处于活化状态[6]。当肿瘤细胞与成纤维细胞共培养时,肿瘤细胞可通过某些细胞因子介导的信号传导激活成纤维细胞并诱导癌症相关成纤维细胞细胞(CAFs)形成[7, 8]。CAFs能分泌胶原蛋白和细胞外基质,还能分泌多种细胞因子,如白细胞介素-6、转化生长因子-β等,促进肿瘤细胞的增殖和侵袭[9]。因此,深入了解TME中CRC细胞与成纤维细胞的相互作用关系有助于开发新的CRC治疗策略,鉴于条件培养基能够提供一个更加可控和特定的环境,我们使用CRC细胞和成纤维细胞的条件培养基,研究某种细胞的分泌成分对另一种细胞的影响。
目前公认的可特异性调控细胞的多种生物化学过程的信号通路有JAK/STAT3、AKT/PI3K、mTOR 和ERK1/2等,与各种肿瘤细胞、基质细胞和细胞因子相互作用有关[9, 10],然而仍没有研究深入揭示正常结直肠成纤维细胞条件培养基对CRC细胞恶性进展的相关信号通路。我们前期研究证明了CRC细胞的条件培养基可通过ERK通路诱导CRC相关CAFs 的形成[11],但结直肠正常成纤维细胞对CRC细胞的影响及特定信号通路尚未见报道。本研究探讨了人结直肠成纤维细胞(CCD-18Co)的条件培养基对CRC 细胞(HCT116,Caco-2)的影响及其具体机制,为开发靶向TME的CRC治疗提供新的思路。
1 材料和方法
1.1 材料
结直肠癌HCT116、Caco-2细胞和人正常结直肠成纤维CCD-18Co细胞(ATCC);1640培养基、胎牛血清、DMEM/F12(Gibco);TRIzol、预染蛋白marker(ThermoFisher);反转录试剂、SYBR qPCR Master Mix(Vazyme);一抗稀释液、特超敏ECL 化学发光底物(Biosharp);青-链霉素、细胞裂解液、BCA 试剂盒和抗α-Tubulin 抗体(Beyotime);B27、FGF、EGF(Ramp;D);抗ERK、p-ERK、AKT、p-AKT、STAT3 和p-STAT3 抗体(Cell Signaling Technology);ERK抑制剂SCH772984(MedChemExpress)。
1.2 方法
1.2.1 人正常结直肠成纤维CCD-18Co的条件培养基制备 取对数生长期的人正常结直肠成纤维CCD-18Co细胞,常规消化、重悬并进行计数(1×106/mL),接种8 mL细胞悬液于10 mm皿中,待细胞生长至60%~70%融合,吸弃旧培养基,以PBS 润洗2 次,更换无血清RPMI 1640培养基,继续培养48 h后收集细胞上清液,3000 r/min离心10 min,留上清,0.22 μm孔径的滤器过滤,分装后即用或冻存于-80 ℃中备用。HCT116 和Caco-2 细胞培养在含10%胎牛血清和1%青链霉素的RPMI 1640完全培养基中。
1.2.2 RT-PCR TRIzol 提取细胞总RNA 并保存于-80 ℃。用逆转录试剂盒取1 μg 总RNA,加入5×qRT预混液4 μL,酶混合液1 μL,补足ddH2O至20 μL,混匀离心后50 ℃ 15 min,85 ℃ 5 s,将RNA逆转录为cDNA。PCR扩增cDNA 1 μL、上游引物1 μL、下游引物1 μL、SYBR qPCR 混合液10 μL、补足ddH2O至20 μL,混匀离心后上机。引物序列由上海生工生物工程有限公司合成,具体序列见表1。以GAPDH为内参照。反应体系20 μL,PCR 扩增条件为:95 ℃ 5 min;95 ℃ 30 s,60 ℃ 30 s,72 ℃ 45 s,39个循环,分析熔解曲线。
1.2.3 Western blotting PBS 清洗细胞后,用细胞裂解液冰上裂解细胞30 min,4 ℃离心20 min,收取上清液,BCA法测定样品蛋白浓度。取10 μg 样品进行SDSPAGE凝胶电泳分离、电转至PVDF膜,5%脱脂奶粉室温封闭1 h,一抗(α-Tubulin、STAT3、ERK、AKT稀释比例 1∶1000,p-STAT3、p-ERK、p-AKT稀释比例1∶2000)4 ℃孵育过夜,TBST 清洗3 次,10 min/次,IgG 二抗(1∶5000)室温孵育1.5 h,TBST 清洗5 次,5 min/次,ECL法显色。以α-Tubulin作为内参照,电泳条带灰度值使用实验室仪器自带软件Image J进行分析,以目的条带灰度值与α-Tubulin条带灰度值之比表示目的蛋白的相对表达量。
1.2.4 实时无标记动态细胞分析技术(RTCA) 使用安捷伦公司的 xCELLigence RTCA eSight仪器及培养板E-Plate 16 进行测量,将完全培养基与条件培养基1∶1混合制备成混合培养基,胰酶消化生长密度且状态良好的对数生长期的CRC细胞制成4×104/mL的细胞悬液,向E-plate16中加入50 μL混合培养基,置于DP检测台上测量基线,再加入50 μL上述细胞悬液,并向各孔补充50 μL对应的条件培养基,对照孔补充无血清的1640培养基,每个处理接种4孔。放入37 ℃、5% CO2的培养箱中培养,每隔15 min检测1次细胞指数,持续72 h,动态检测对照组和实验组的细胞增殖指数。
1.2.5 平板克隆实验 取对数生长期的CRC 细胞500/孔,贴壁后加入相应的混合培养基,每组设3 个复孔。2 d/次换液,培养14 d后弃去培养基,用PBS清洗2次,再用4%多聚甲醛固定15 min,弃去固定液,加入结晶紫染色15 min后,回收结晶紫并清洗干净,常温晾干后拍照。
1.2.6 Transwell实验 胰酶消化生长密度合适且状态良好的对数生长期的CRC细胞,用无血清RPMI 1640 培养基重悬,制成细胞悬液,向8 μm 孔径的Transwell小室中加入100 μL 的上述细胞悬液,在下室中加入700 μL/孔CCD-18Co-CM或对照组培养基,每组3个复孔,确保上下室间无气泡,将培养皿放入细胞培养箱中,24 h后弃掉上下室内的培养基,PBS清洗小室两遍,加入4%多聚甲醛溶液固定15~30 min,弃去固定液加入结晶紫溶液染色15 min,用棉棒轻柔地擦掉小室内侧未迁移的细胞,再次清洗小室直至背景清晰,室温自然晾干。显微镜下观察拍照并随机记录不少于5 个视野下的细胞数。
1.2.7 肿瘤球形成实验 CRC细胞在DMEM/F12(1∶1)+1×B27+20 ng/mL FGF+20 ng/mL EGF培养基中培养,加入CCD-18Co-CM。然后将细胞以5000/孔的密度接种到超低附24孔板中。在整个实验过程中,2 d/次更换新鲜培养基。使用无血清培养基形成CRC细胞的肿瘤球体,7 d后消化并传代至第2代再传代7 d。通过光学显微镜观察肿瘤球体的形成并拍照。
1.2.8 ERK 抑制剂处理细胞 在发现CCD-18Co-CM能激活CRC 细胞ERK 信号通路后,ERK 抑制剂SCH772984 处理细胞,再次进行RTCA,克隆形成和Transwell 等表型实验。将人HCT116 和Caco-2 CRC细胞分为对照组(完全培养基与无血清1640培养基1∶1混合后处理)、CCD-18Co-CM组(完全培养基和CCD-18Co细胞条件培养基1∶1混合后处理)、iERK组(对照组混合培养基+100 nmol/L 的SCH772984 处理)和CCD-18Co-CM+iERK组(CCD-18Co-CM组混合培养基+100 nmol/L SCH772984 处理)。SCH772984 需提前加入CRC细胞中预处理2 h后再加入混合培养基,未添加SCH772984 的其他处理组需补充相应剂量的DMSO,每组处理时间为72 h。
1.3 统计学分析
所有实验重复至少3次,数据经检验符合正态分布且具有方差齐性,以均数±标准差表示。用SPSS 23软件对所有实验数据进行统计学分析,组间比较采用t检验,以Plt;0.05为差异有统计学意义。
2 结果
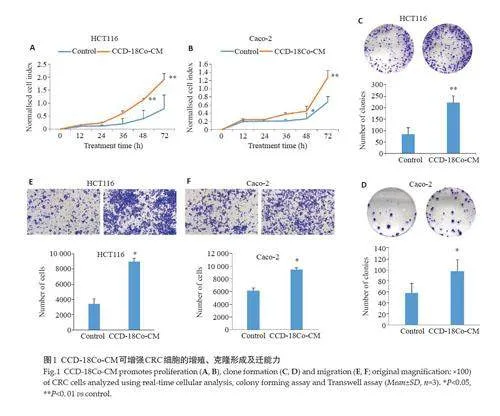
2.1 CCD-18Co-CM增强CRC细胞增殖、克隆形成及迁移能力
RTCA、克隆形成及Transwell 实验检测发现处理组HCT116和Caco-2细胞的增殖、克隆形成及迁移能力均显著增强(图1A~F,Plt;0.05)。
2.2 CCD-18Co-CM促进CRC细胞的干性特征
CD29、CD44、CD166、MYC 和EPCAM的mRNA在CCD-18Co-CM处理的HCT116 和Caco-2 细胞中表达水平升高(图2A、B,Plt;0.05)。同时肿瘤球形成实验也证明CCD-18Co-CM处理后的HCT116 和Caco-2 细胞成球能力增强(图2C、D)。
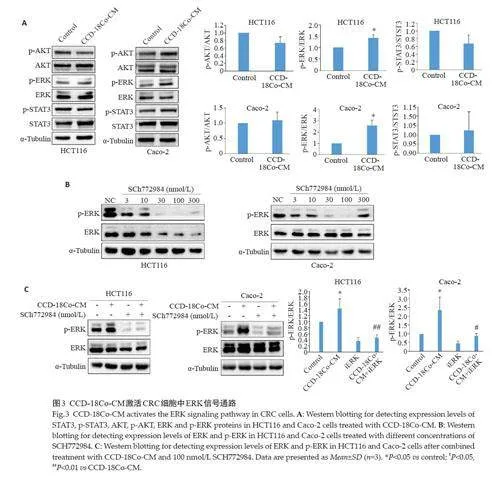
2.3 CCD-18Co-CM激活CRC细胞中ERK信号通路
CCD-18Co-CM可激活HCT116 和Caco-2 细胞的ERK信号通路,ATK和STAT3 通路未见激活(图3A,Plt;0.05)。应用ERK通路抑制剂SCH772984(iERK)处理CRC细胞,筛选出HCT116和Caco-2细胞中iERK的抑制浓度为100 nmol/L(图3B),iERK 可显著抑制CCD-18Co-CM对HCT116和Caco-2细胞ERK通路的激活作用(图3C,Plt;0.05)。
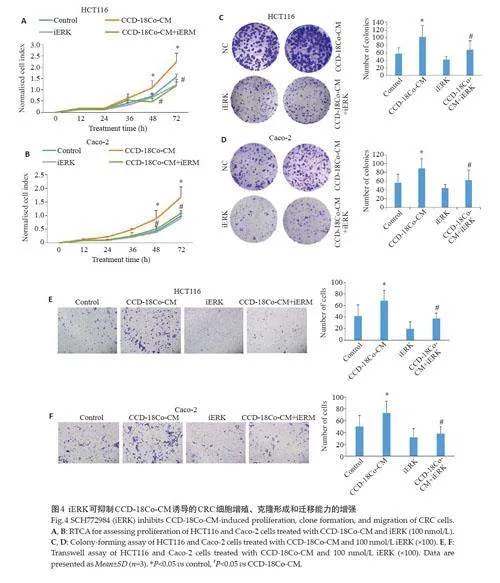
2.4 iERK可抑制CCD-18Co-CM诱导的CRC 细胞增殖、克隆形成和迁移能力的增强
CCD-18Co-CM诱导的HCT116 和Caco-2 细胞增殖、克隆形成及迁移能力的增强可被iERK显著抑制(图4A~F,Plt;0.05)。
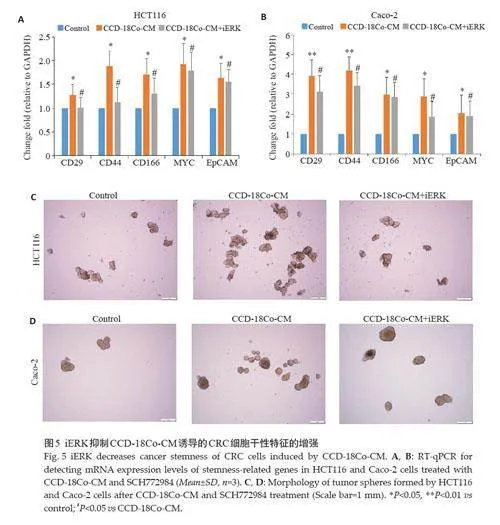
2.5 iERK抑制CCD-18Co-CM诱导的CRC细胞干性特征的增强
iERK降低了CCD-18Co-CM诱导的CRC细胞中CD29、CD44、CD166、MYC和EPCAM的表达水平(图5A、B,Plt;0.05)。肿瘤球形成实验表明,iERK降低了CCD-18Co-CM诱导的CRC细胞的球形能力(图5C、D)。
3 讨论
根据预测,至2040 年全球CRC 新病例数将达到320万,且发病年龄呈低龄化趋势[2]。CRC的治疗手段包括手术切除、放化疗、免疫治疗、靶向治疗等多种治疗方式,但其致死率仍高达癌症死亡原因的9.4%,且伴随易耐药、转移、复发与预后差等特点,因此开发新的CRC治疗方案尤为重要。TME是CRC发展、生长和转移的内部和外部环境,它由多种控制癌症进展的细胞和细胞因子组成,因此从TME入手,寻找和发现CRC治疗策略是近年来CRC治疗新的路径。本文研究TME中分布最广泛的人结肠成纤维细胞对CRC细胞的影响及其机制,为CRC治疗提供思路。
正常组织中的成纤维细胞来自于细胞外基质(ECM)内的静息间充质细胞,可在伤口愈合、组织炎症和纤维化时被激活,以促进修复和再生[12]。有研究证明,成纤维细胞可通过重编程树突状细胞和单核细胞使其发生功能障碍或者转变成免疫抑制型细胞,导致TME中免疫抑制状态的形成,促进乳腺癌肺转移的发生[13]。在CRC中研究成纤维细胞与CRC细胞相互作用关系时,Chen等[14]研究认为,与正常成纤维细胞共培养后,CRC细胞的增殖,迁移和侵袭能力增强,而Ca2+/钙调蛋白-依赖性蛋白激酶 II (CaMKII)敲除的正常成纤维细胞不具有增强CRC 细胞恶性能力的作用,提示CaMKII是成纤维细胞与CRC细胞相互交流的关键分子。本研究用正常人结肠成纤维细胞条件培养基CCD-18Co-CM处理CRC细胞,证明了同样具有增强CRC细胞增殖、克隆形成及迁移能力的功能。
研究证实肿瘤干细胞(CSCs)是CRC复发、转移及恶性进展的关键原因之一,常见的CSCs细胞表面分子标志物有CD24、CD29、CD44、CD133、CD166、ALDH1、EPCM等[15, 16]。有研究提示,隐层肌成纤维细胞不但是干细胞组成部分,也是CAFs 的重要组成部分[17]。本研究结果显示,CCD-18Co-CM处理CRC细胞,其成球能力增强,干性分子标志物表达增加,提示肿瘤微环境中正常成纤维细胞可增强CRC细胞的恶性特征。
我们前期研究结果证明,CRC细胞条件培养基诱导活化的CCD-18Co细胞表达典型CAFs特征,其相关分子标志物如TGF-β、α-SMA、FAP和FN表达增加,增殖及运动能力增强,提示CRC-CM可诱导正常成纤维细胞向典型CAFs 表型特征转变[11]。关于CRC诱导正常成纤维细胞向CAFs 的转化近年来已有报道[18-20],活化后的CAFs可生成大量生长因子和ECM调节炎症和免疫,肿瘤细胞则利用其分泌的促生长因子和ECM改变,实现自身地快速生长和远处迁移、侵袭。CAFs能够执行自分泌、细胞外基质重塑和动态免疫调节任务[21],它们产生限制DC细胞成熟和细胞毒性T细胞活性的物质,促进Treg细胞和骨髓源性抑制细胞分化。CAFs还可以刺激上皮细胞中的上皮-间质转化,提高肿瘤细胞的治疗抵抗力,其代谢产物可以为肿瘤细胞提供营养支持[22]。在炎症环境中,细胞因子如IL-1、IL-17 和IL-11是强大的成纤维细胞刺激因子,可极大地激活成纤维细胞[23]。也有文献报道,成纤维细胞与癌细胞通过JNKIL-1-CXCL9/10-CXCR3 信号轴进行的信号交流是乳腺癌肺转移的重要机制[24]。结合本研究和前期报道,提示在CRC TME中,CRC细胞与成纤维细胞相互作用,促进CRC细胞的恶性进展。
本研究同时证明,CCD-18Co-CM激活CRC细胞中ERK信号通路,抑制ERK信号通路可以显著降低CCD-18Co-CM诱导的HCT116 和Caco-2 细胞的恶性表型和干性特征,ERK 抑制剂SCH772984 可逆转CCD-18Co 细胞对CRC细胞的作用,这意味着人结肠直肠成纤维细胞可能通过ERK信号途径参与CRC细胞的恶性进展。CAFs分泌的成分通过ERK信号通路促进多种肿瘤的恶性进展,其中Periostin 可通过ERK通路促进非小细胞肺癌的增殖和药物抵抗[25],u-PA可通路ERK通路促进食管鳞状细胞癌的恶性进展[26],HMGB1可通过ERK途径诱导乳腺癌细胞自噬和化疗抵抗[27],我们前期研究也证明SCH772984降低CAFs相关标志物的表达水平并抑制CCD-18Co细胞增殖、克隆形成和迁移[11]。目前针对ERK信号通路开发的小分子抑制剂治疗CRC的有效性和作用机制不断被发现[28-30],我们的发现证明了ERK信号通路是CRC TME中成纤维细胞和CRC相互作用的主要通路,但其具有的激活机制未进一步探讨,未来应深入探索ERK信号通路在体内的具体作用机制,将ERK抑制剂应用于临床前CRC动物模型中,为ERK抑制剂成为治疗CRC奠定基础。
因此,结直肠正常成纤维细胞通过激活ERK信号通路来促进CRC的恶性进展。应用ERK抑制剂阻断肿瘤细胞和正常成纤维细胞相互作用可能是CRC治疗中靶向TME的一种有前景的策略。
参考文献:
[1] Dyba T, Randi G, Bray F, et al. The European cancer burden in 2020:
incidence and mortality estimates for 40 countries and 25 major
cancers[J]. Eur J Cancer, 2021, 157: 308-47.
[2] Arnold M, Sierra MS, Laversanne M, et al. Global patterns and
trends in colorectal cancer incidence and mortality[J]. Gut, 2017, 66
(4): 683-91.
[3] Mao XQ, Xu J, Wang W, et al. Crosstalk between cancer-associated
fibroblasts and immune cells in the tumor microenvironment: new
findings and future perspectives[J]. Mol Cancer, 2021, 20(1): 131.
[4] Bejarano L, Jordāo MJC, Joyce JA. Therapeutic targeting of the
tumor microenvironment[J]. Cancer Discov, 2021, 11(4): 933-59.
[5] Tiwari A, Trivedi R, Lin SY. Tumor microenvironment: barrier or
opportunity towards effective cancer therapy[J]. J Biomed Sci,
2022, 29(1): 83.
[6] Verginadis II, Avgousti H, Monslow J, et al. A stromal Integrated
Stress Response activates perivascular cancer-associated fibroblasts
to drive angiogenesis and tumour progression[J]. Nat Cell Biol,
2022, 24(6): 940-53.
[7] Sahai E, Astsaturov I, Cukierman E, et al. A framework for
advancing our understanding of cancer-associated fibroblasts[J].
Nat Rev Cancer, 2020, 20(3): 174-86.
[8] Fang T, Lv HW, Lv GS, et al. Tumor-derived exosomal miR-1247-
3p induces cancer-associated fibroblast activation to foster lung
metastasis of liver cancer[J]. Nat Commun, 2018, 9(1): 191.
[9] Chen MH, Gu YY, Zhang AL, et al. Biological effects and
mechanisms of matrine and other constituents of Sophora flavescens
in colorectal cancer[J]. Pharmacol Res, 2021, 171: 105778.
[10]Rausio H, Cervera A, Heuser VD, et al. PIK3R1 fusion drives
chemoresistance in ovarian cancer by activating ERK1/2 and
inducing rod and ring-like structures[J]. Neoplasia, 2024, 51:
100987.
[11]邓 婷, 杜伯雨, 郗雪艳. 结直肠癌细胞通过激活成纤维细胞的ERK
通路诱导癌症相关成纤维细胞的形成[J]. 南方医科大学学报,
2023, 43(6): 943-51.
[12]Correa-Gallegos D, Jiang DS, Rinkevich Y. Fibroblasts as
confederates of the immune system[J]. Immunol Rev, 2021, 302(1):
147-62.
[13]Gong Z, Li Q, Shi JY, et al. Lung fibroblasts facilitate pre-metastatic
niche formation by remodeling the local immune microenvironment
[J]. Immunity, 2022, 55(8): 1483-500. e9.
[14]Chen W, Chen YW, Su J, et al. CaMKII mediates TGFβ1-induced
fibroblasts activation and its cross talk with colon cancer cells[J].
Dig Dis Sci, 2022, 67(1): 134-45.
[15]Najafi M, Farhood B, Mortezaee K. Cancer stem cells (CSCs) in
cancer progression and therapy[J]. J Cell Physiol, 2019, 234(6):
8381-95.
[16]Angius A, Scanu AM, Arru C, et al. Portrait of cancer stem cells on
colorectal cancer: molecular biomarkers, signaling pathways and
miRNAome[J]. Int J Mol Sci, 2021, 22(4): 1603.
[17]Ferrer-Mayorga G, Niell N, Cantero R, et al. Vitamin D and Wnt3A
have additive and partially overlapping modulatory effects on gene
expression and phenotype in human colon fibroblasts[J]. Sci Rep,
2019, 9(1): 8085.
[18]Dias Carvalho P, Mendonça S, Martins F, et al. Modulation of
fibroblast phenotype by colorectal cancer cell-secreted factors is
mostly independent of oncogenic KRAS[J]. Cells, 2022, 11(16):
2490.
[19]Hassametto A, Tanomrat R, Muangthong T, et al. Role of oxidative
stress-dependent C/EBPβ expression on CAF transformation
inducing HCT116 colorectal cancer cell progression; migration and
invasion[J]. Asian Pac J Cancer Prev, 2023, 24(11): 3825-35.
[20]Si GF, Li SY, Zheng Q, et al. MiR-1246 shuttling from fibroblasts
promotes colorectal cancer cell migration[J]. Neoplasma, 2021, 68
(2): 317-24.
[21]Yoshida GJ. Regulation of heterogeneous cancer-associated
fibroblasts: the molecular pathology of activated signaling pathways
[J]. J Exp Clin Cancer Res, 2020, 39(1): 112.
[22]Mhaidly R, Mechta-Grigoriou F. Role of cancer-associated
fibroblast subpopulations in immune infiltration, as a new means of
treatment in cancer[J]. Immunol Rev, 2021, 302(1): 259-72.
[23]Widjaja AA, Chothani S, Viswanathan S, et al. IL11 stimulates IL33
expression and proinflammatory fibroblast activation across tissues
[J]. Int J Mol Sci, 2022, 23(16): 8900.
[24]Pein M, Insua-Rodríguez J, Hongu T, et al. Metastasis-initiating
cells induce and exploit a fibroblast niche to fuel malignant
colonization of the lungs[J]. Nat Commun, 2020, 11(1): 1494.
[25]Takatsu F, Suzawa K, Tomida S, et al. Periostin secreted by cancerassociated
fibroblasts promotes cancer progression and drug
resistance in non-small cell lung cancer[J]. J Mol Med, 2023, 101
(12): 1603-14.
[26]Tian BQ, Chen XJ, Zhang HH, et al. Urokinase plasminogen
activator secreted by cancer-associated fibroblasts induces tumor
progression via PI3K/AKT and ERK signaling in esophageal
squamous cell carcinoma[J]. Oncotarget, 2017, 8(26): 42300-13.
[27]Liu L, Liu SC, Luo HJ, et al. GPR30-mediated HMGB1
upregulation in CAFs induces autophagy and tamoxifen resistance
in ERα‑positive breast cancer cells[J]. Aging, 2021, 13(12):
16178-97.
[28]Tayama H, Karasawa H, Yamamura A, et al. The association
between ERK inhibitor sensitivity and molecular characteristics in
colorectal cancer[J]. Biochem Biophys Res Commun, 2021, 560:
59-65.
[29]Lin K, Zhao Y, Tang YQ, et al. Collagen I-induced VCAN/ERK
signaling and PARP1/ZEB1-mediated metastasis facilitate OSBPL2
defect to promote colorectal cancer progression[J]. Cell Death Dis,
2024, 15(1): 85.
[30]Liu W, Tang JT, Gao W, et al. PPP2R1B abolishes colorectal cancer
liver metastasis and sensitizes Oxaliplatin by inhibiting MAPK/ERK
signaling pathway[J]. Cancer Cell Int, 2024, 24(1): 90.
(编辑:经 媛)
猜你喜欢
世界科学技术-中医药现代化(2021年5期)2021-11-05 06:56:30
方圆(2017年12期)2017-07-17 17:48:12
中国医药导报(2016年28期)2017-01-06 19:55:33
中国医药导报(2016年29期)2016-12-27 18:02:30
中国民族民间医药·上半月(2016年11期)2016-12-26 13:59:46
中国现代医生(2016年27期)2016-12-21 16:20:20
中国实用医药(2016年28期)2016-12-07 22:26:23
中国实用医药(2016年27期)2016-11-30 12:06:21
中国实用医药(2016年27期)2016-11-30 10:51:38
医学信息(2016年29期)2016-11-28 10:20:58
