
云南十里香茶树空间转录组测序研究
2024-07-10 06:00王冬雪满佳旭武思敏赵雪婷张冬英
茶叶科学 2024年3期
关键词:差异基因
王冬雪 满佳旭 武思敏 赵雪婷 张冬英
摘要:近年来,云南大叶种茶和古茶树资源备受关注,而对于云南小叶种茶树资源研究报道较少。十里香茶树品种是云南特有的小叶种茶树资源,品质独特且饮用历史悠久。空间转录组技术作为一种新兴的基因表达分析技术,目前尚未见在茶树资源应用上的文献报道。利用空间转录组测序技术对十里香茶树嫩芽的基因表征情况和空间调控机制进行研究,结果显示,Spot聚类分析识别嫩芽细胞类型,划分为13个不同细胞类型cluster,构建空间转录组图谱,观察到不同细胞类型cluster在嫩芽的两种发育时期的空间表达位置存在差异,呈现空间异质性。进一步鉴定不同细胞类型cluster中的差异基因,主要以抗逆胁迫、生长发育调控为主,抗逆胁迫代表性基因为LOC114312694、LOC114319171、LOC114320792、LOC114287723、LOC114284011、LOC114289235,生长发育代表性基因为LOC114263486、LOC114320821、LOC114292779、LOC114321117、LOC114286858;并绘制空间分布图,发现这些抗逆胁迫和生长发育基因在幼叶中高表达,这说明在嫩芽发育的早期阶段,其在抗逆胁迫和生长发育调控方面发挥重要作用。GO与KEGG富集分析发现,十里香茶树嫩芽的差异基因涉及多个重要通路,如翻译、茉莉酸信号调控、钙离子结合、植物激素信号转导等,这些都与茶树生长发育紧密相关。此研究结果可为十里香茶树发育生物学提供一定的科学依据,同时也为其他茶树资源的研究提供一种新的思路。
关键词:十里香;空间转录组;差异基因;空间异质性;发育生物学
中图分类号:S571.1;S326 文献标识码:A 文章编号:1000-369X(2024)03-399-12
Spatial Transcriptome Sequencing of Shilixiang in Yunnan Province
WANG Dongxue1,2, MAN Jiaxu3, WU Simin1,2, ZHAO Xueting1, ZHANG Dongying1,2*
1. College of Science, Yunnan Agricultural University, Kunming 650201, China;
2. Key Laboratory of Pu'er Tea Science, Yunnan Agricultural University, Kunming 650201, China;
3. Institute of Agricultural Products Processing, Yunnan Academy of Agricultural Sciences, Kunming 650201, China
Abstract: In recent years, Yunnan's large leaf tea and ancient tea resources have attracted much attention, while there are relatively few reports on the research of small leaf tea resources. Shilixiang, a distinctive small leaf tea resource in Yunnan, possessed unique quality and a long drinking history. Spatial transcriptome technology, an emerging gene expression analysis technique, has not been previously applied to tea resources according to current literature. The gene characterization and spatial regulation mechanism of the tender buds of Shilixiang were researched by spatial transcriptome sequencing technology in this study. The results show that 13 clusters of different cell types in the tender bud cells were identified by a spot clustering analysis and the spatial transcriptome map was constructed. The expression positions of clusters during the two developmental stages of the bud were different and spatial heterogeneity was observed from this analysis. Further exploration involved the identification of differential genes in various cell type clusters, with a focus on stress response and growth and development regulation. Representative stress responsive genes included LOC114312694, LOC114319171, LOC114320792, LOC114287723, LOC114284011 and LOC114289235. Meanwhile, representative growth and development genes included LOC114263486, LOC114320821, LOC114292779, LOC114321117, and LOC114286858. A spatial distribution map illustrated the high expression of these stress response and growth development genes in young leaves, indicating their crucial role in the early stage of tender bud development. Further GO and KEGG enrichment analysis reveal that the differential genes in the tender buds of Shilixiang are associated with multiple important pathways. These pathways included translation, jasmonic acid signal regulation, calcium ion binding, and plant hormone signal transduction, all of which are closely linked to the growth and development of tea plants. The results of this study provided a solid scientific foundation for understanding the developmental biology of Shilixiang. Additionally, they provided a new perspective for exploring other tea resources.
Keywords: Shilixiang, spatial transcriptome, differential gene, spatial heterogeneity, developmental biology
随着科学技术的日新月异,茶树基因组的研究从未止步[1]。自2010年中国科学院启动“茶基因组”计划以来,历经7年,构建了第一个茶树基因组(阿萨姆种基因组),为后续的茶树研究提供了先例[2]。茶树基因组具有高杂合性,极大制约了茶树基因组的进展[3-4]。迄今为止,在茶树基因组数据库中,已经公布众多茶树基因组,如云抗10号[2]、舒茶早[5-6]、龙井43[7]等。这些基因组的公布为茶树的遗传育种、进化起源等提供了一定的科学参考[8]。目前,转录组学分析已被广泛用于揭示茶树特征性次级代谢通路、抗逆性和产量相关的转录调控等机制的研究[9-11],包括采用不同组织的不同时空样本[12]、生物胁迫样本和非生物胁迫样本[13-14]。也有研究者使用二代转录组测序技术对茶树舒茶早品种进行了转录组测序[15],这使得茶树二代转录组测序研究进入到快速发展阶段。近些年来,大量的基因组测序加速了茶树的遗传育种进程[16-19],这些研究结果对促进茶树品种改良具有重要意义。
空间转录组技术作为近几年新兴的高通量测序技术,为我们解析基因在组织结构中的复杂调控提供了一种全新的视角。传统的转录组技术,虽可以提供基因在总体水平上的表达信息,但不能揭示基因在组织或细胞水平上的具体分布情况。而空间转录组技术可在组织原位同时获得基因表达特征和空间分布数据,进一步推进了对组织原位细胞真实基因表达的研究。拟南芥是第一个构建空间转录组图谱的样本,该图谱揭示了拟南芥跨组织结构域的141个差异表达基因[20]。在马齿苋植物中应用空间转录组技术聚焦光合作用场所的叶肉细胞和束鞘细胞群体,进行了两种不同的空间基因表达分析[21]。通过构建兰花发育过程中的空间转录组技术图谱,为进一步研究兰花开花过程中复杂而重要的基因调控网络提供了宝贵的资源[12]。也有研究报道,利用空间转录组技术创建的番茄愈伤组织的细胞图谱,可以揭示其异质性并鉴定出不同的细胞类型[22]。在江南卷柏(Selaginella moellendorffii)根系中进行空间转录组测序研究,支持了控制根系发育机制高度趋同进化的观点[23]。目前,空间转录组技术在茶树资源中的应用还未见报道。
云南是茶树的起源地,大叶种茶树资源和古茶树资源备受人们关注[25],而当地小叶种茶树资源关注度较低。十里香茶树作为云南特有的小叶种茶树资源,栽培历史悠久,其最早应用可追溯到唐代[25]。近些年来,十里香茶树研究多集中在遗传多样性和种质资源保护、栽培技术、新品种的选育和产量优化等方面,且多采用传统研究技术,目前整体研究水平较低,研究进程较为缓慢[26-27]。本文利用空间转录组技术对十里香茶树嫩芽进行研究,旨在为十里香茶树发育生物学提供科学依据,同时也为其他茶树资源的研究提供一种新的思路。
1 材料与方法
1.1 试验材料
样品于2022年6月采集自昆明市云南农业大学茶叶种植基地,茶树品种为十里香,同一茶树上取5个嫩芽。
1.2 样品制备
使用异戊烷和液氮浴冷冻新鲜嫩芽,用OCT包埋冷冻组织样本,﹣80 ℃保存;在冷冻切片机中进行冷冻切片;并收集组织切片用于RNA提取及质检,保证组织冷冻过程中RNA无降解。
1.3 组织优化
将冷冻切片按照区域贴在组织优化的玻片上,然后对组织切片进行甲醇固定、苏木精-伊红染色(Hematoxylin and eosin staining,HE染色)和明场成像,再进行不同时间梯度透化和荧光标记的cDNA合成,最后移除组织进行荧光成像,通过荧光信号的强度和弥散程度确定最佳的透化条件,即荧光信号的强度最大且没有弥散为最佳透化时间,用于后续基因表达文库的构建。
1.4 基因表达文库构建和质检
将组织切片贴于基因表达的玻片上,进行甲醇固定、H&E染色和明场成像;根据组织优化确定的透化时间进行组织透化,基因表达玻片上的组织切片释放的mRNA被spot上的特殊引物捕获并逆转录成cDNA。从载玻片上收集带有空间条形码的cDNA,经过二链合成、变性、PCR扩增得到初步cDNA,再通过酶切片段化处理、末端修复加A尾、磁珠片段筛选、接头连接、磁珠纯化、添加样本indexes PCR构建标准二代测序文库。
1.5 组织透化和cDNA合成
根据组织优化试验确定的透化时间进行组织透化,使细胞中的mRNA得到释放,并结合到相应的捕获探针上;再进行mRNA逆转录,得到完整cDNA一链;经过二链合成、二链变性回收、cDNA扩增和cDNA纯化得到完整的cDNA。
1.6 cDNA定量及质控
取1 ?L样品稀释至2 ng·?L-1,使用Qubit测定纯化后cDNA产物浓度,使用High sensitivity Agilent Technologies 2100 Bioanalyzerd对cDNA产物峰型进行测定。
1.7 文库定量和质控
取10 ?L cDNA进行建库,通过片段化、末端修复加A尾,接头连接,磁珠纯化,样本index PCR,PCR后的磁珠双端片段筛选等步骤完成文库构建。并利用Qubit4.0测定文库浓度;稀释到合适的浓度,利用Qseq400进行片段检测,一般文库分布于在300~800 bp,平均片段分布于400~500 bp。
1.8 上机测序
文库质检合格后,利用百迈客公司二代测序仪平台对空间基因表达文库进行测序,测序策略为PE150。
1.9 测序数据及其质量评估
采用百迈客公司Illumina技术对样本的转录组进行双端150 bp测序。为确保测序数据的质量和可靠性,使用Fastp(v0.23.4)对数据进行预处理,去除低质量序列,限制N碱基的存在,修剪了序列的前端和尾端,确保所保留的序列长度不低于75 bp。
1.10 BSTMarits分析
使用百迈客公司平台的BSTMatrix数据分析软件包,以过滤后的fastq文件和组织切片的甲苯胺蓝染色图片为输入数据,进行参考基因组比对、组织检测和Spatial Barcode/UMI统计,生成spot-基因表达矩阵。
1.11 Spot聚类分析
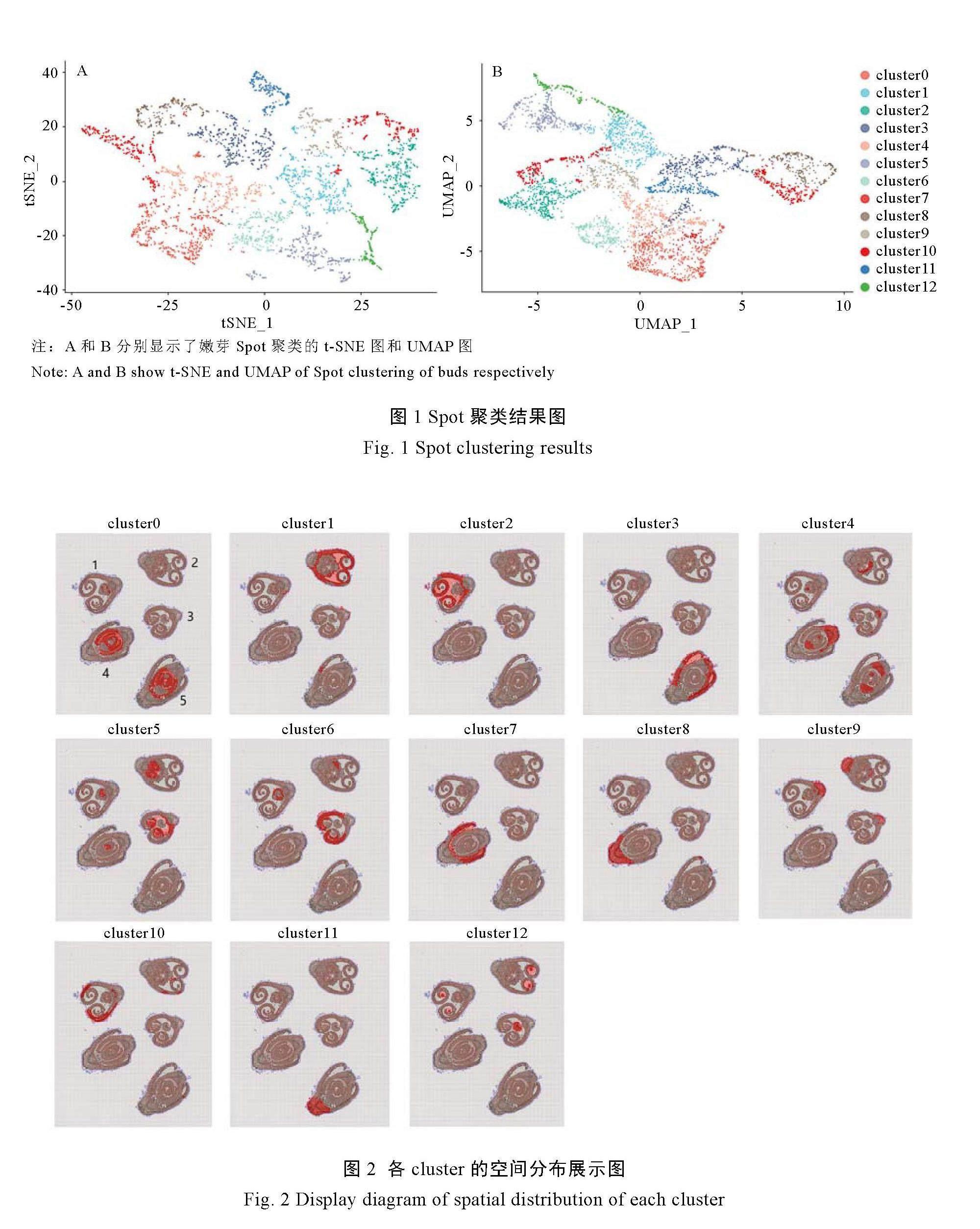
利用Seurat包中的sctransform算法对数据进行标准化处理,之后通过vst算法筛选出数据中表达变化最大的基因集,默认选择3 000个差异基因作为高变异基因(High-variance genes,HVGs),使用UMAP[28]和t-SNE[29]两种降维手段进行spot聚类分析,将空间spot划分成不同区域cluster并进行后续分析。
1.12 区域功能富集分析
使用7个公共数据库根据差异基因序列的相似性进行比对注释,包括Nr非冗余蛋白质数据库(www.ncbi.nlm.nih.gov)、Swiss-Prot蛋白数据库(www.expasy.org)、GO基因本体数据库(http://geneontology.org)、COG同源蛋白簇数据库(www.ncbi.nlm.nih.gov/research/
cog)、KOG真核同源基团数据库(http://genome.
jgi-psf.org/help/kogbrowser.jsf)、Pfam大型蛋白结构域家族的数据库(http://pfam.xfam.org)和京都基因与基因组百科全书(KEGG,www.genome.jp/kegg),得到基因的注释信息,并为对应cluser进行空间转录组亚群分析、差异表达基因分析、GO/KEGG功能富集分析。
2 结果与分析
2.1 数据质量评估结果
由于目前还没有十里香茶树基因组数据作为参考,因此将十里香茶树测序数据与数据库内已有的茶树基因组进行比对,发现与舒茶早基因组数据的比对率最高,达到80%以上。转录组比对结果同样显示与舒茶早比对率最高。而且舒茶早是中小叶品种,因此选择舒茶早作为参考基因组。本研究共获得59.42 G的原始测序数据,统计获取352 196个Spots,去除低质量的序列后获得198 075 303个reads。十里香茶树基因组的GC含量值为35.32%;质量值≥20的碱基所占的百分比(Q20)为93.39%,质量值≥30的碱基所占的百分比(Q30)为87.75%,与参考基因组和转录组的平均比对率分别为93.52%和77.22%,测序饱和度为71.38%,测序各项指标质量较好,可以满足后续分析条件。
2.2 十里香茶树嫩芽空间转录组图谱的构建
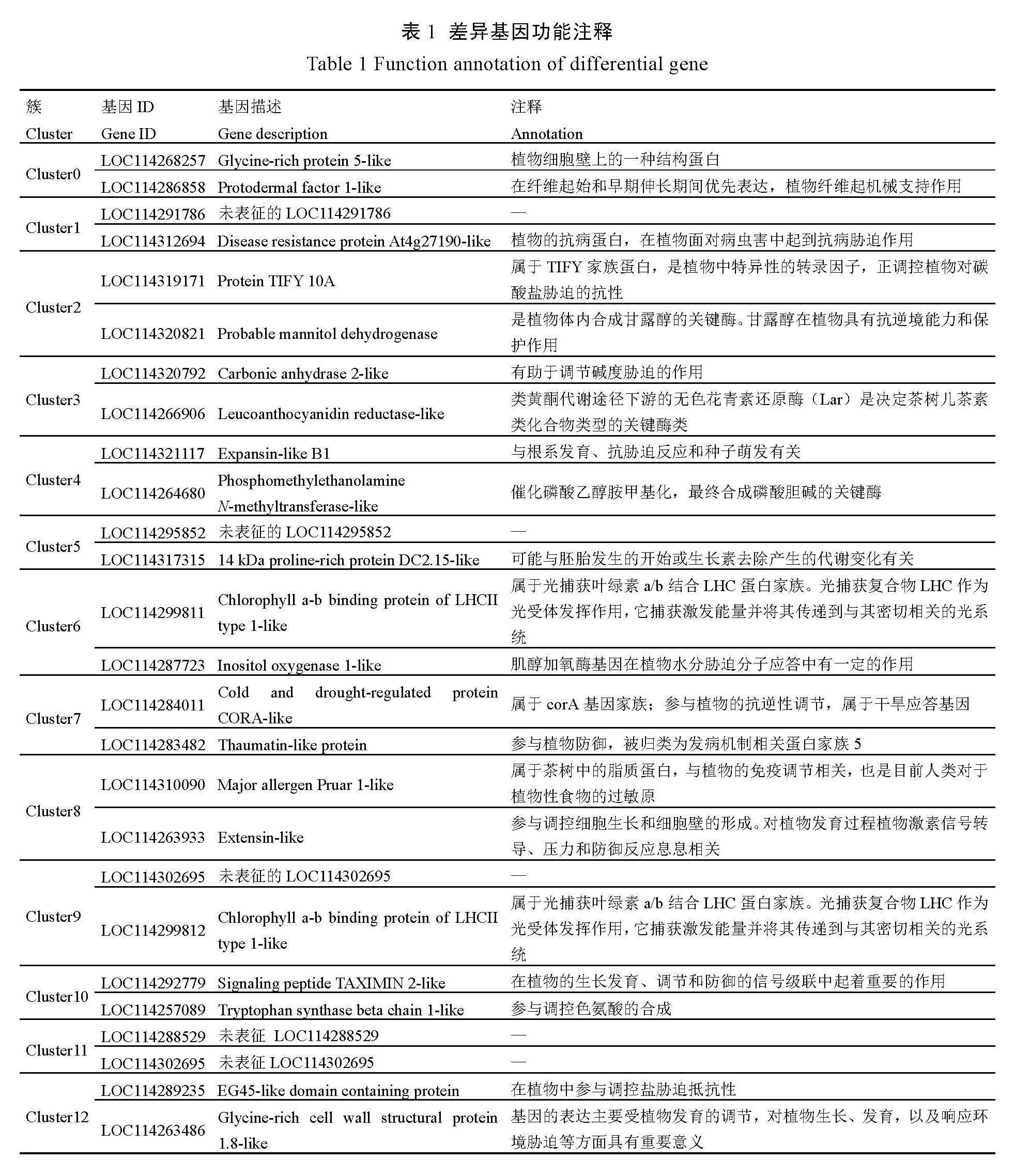
聚类分析获得不同细胞类型的cluster共13个,不同颜色的区域代表不同功能,空间距离代表表达模式的关联程度(图1)。构建不同细胞类型cluster分布空间转录组图谱,可以观察细胞群的全局性和局部相似性。从图2中可以看到两种发育形态的嫩芽(嫩芽主要包括幼叶和芽轴),第一种嫩芽的发育形态(以cluster0的123为例),cluster4、cluster5、cluster9主要富集于芽轴;cluster1、cluster2、cluster6、cluster10富集于幼叶。第二种嫩芽发育形态(以cluster0的45为例),cluster3、cluster7富集于幼叶;cluster4、cluster8、cluster11富集于芽轴。这一分析发现,不同细胞类型cluster在不同的发育阶段,基因的表达位置存在空间异质性,暗示嫩芽发育过程中基因的表达呈动态变化。准确划分细胞边界和鉴定细胞的确切位置对理解嫩芽区域特异性的细胞分化过程至关重要。
2.3 十里香茶树差异基因的表达分析
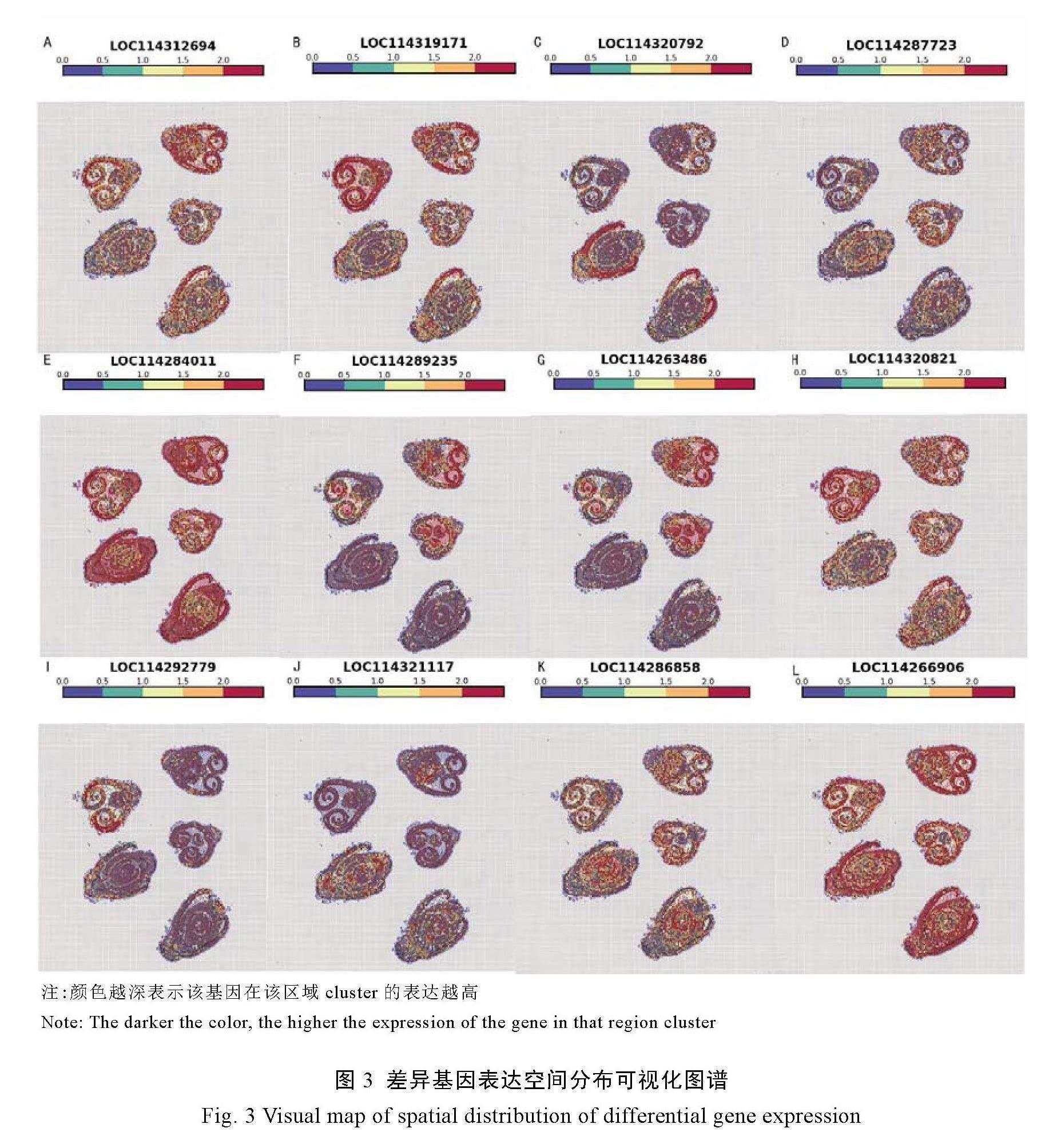
对十里香茶树嫩芽中不同细胞类型cluster的差异基因进行功能注释,由表1可知,这些基因主要涉及植物抗逆性调节、生长发育以及儿茶素合成。从表1中选取具有代表性基因进一步绘制空间转录组图谱,如图3所示。LOC114312694、LOC114319171、LOC114320792、LOC114287723、LOC114284011和LOC114289235等基因参与了植物生长发育中的抗逆性调节过程,这些基因红色基本集中在幼叶区域,在幼叶中有高表达(图3A~图3F)。幼叶生长在植物的顶端和外端,对环境变化非常敏感,在生长发育过程中受到各种环境压力和逆境条件的挑战,从而演化出一种适应机制,故抗逆性基因表达较高。LOC114263486、LOC114320821、LOC114292779、LOC114321117、LOC114286858等基因参与调控生长发育相关,红色基本集中在幼叶区域(图3G~图3K),说明这些与调控生长发育相关的基因在幼叶
区域有高表达,在芽轴表达较低。LOC1142266906是类黄酮代谢途径下游的无色花青素还原酶,它是决定茶树儿茶素类化合物类型的关键酶类,具有多种重要的生物学功能。如图3L所示,LOC1142266906基因在整个嫩芽组织高表达,说明类黄酮代谢在十里香茶树嫩芽发育中发挥重要的作用。
2.4 十里香茶树嫩芽差异基因GO功能分类注释
GO功能分类注释结果分为生物学过程、细胞组分和分子功能3类(图4)。生物学过程主要包括生物调节(32%)、对刺激的反应(29%)、代谢过程(18%)、细胞过程(16.4%)
等。细胞组分中细胞部分、细胞器、细胞器部分和大分子复合物分别占比为65%、6.4%、7.7%、8.8%。分子功能中,酶调节活性、结合活性、蛋白活性占比较高,分别为20.8%、26.7%、23.4%。
从GO富集结果选取每个cluster最显著的GO通路富集项(表2),结合2.3章节分析发现,抗逆胁迫基因聚类于cluster1、cluster2、cluster4、cluster6、cluster7和cluster12,其在GO富集通路主要涉及翻译、核糖体、核小体、细胞壁大分子分解代谢、茉莉酸介导信号通路调控、几丁质分解代谢、蛋白质异源二聚化活性、叶绿体类囊体膜和光系统Ⅰ、光系统Ⅱ等。生长发育基因聚类于cluster0、cluster2、cluster4和cluster10。GO富集通路主要关于核糖体、细胞壁、核小体、转录辅阻遏蛋白活性、转录共调节因子活性、钙离子结合、对刺激的反应调控、茉莉酸介导信号通路调控、细胞壁大分子分解代谢、几丁质分解代谢、蛋白质异源二聚化活性、叶绿体类囊体膜和光系统Ⅰ、
光系统Ⅱ等通路。翻译、核糖体和核小体是植物细胞内生命活动的关键组成部分,直接参与蛋白质合成,而蛋白质是植物嫩芽发育和生理过程中不可或缺的重要分子。细胞壁大分子分解代谢、茉莉酸介导信号通路调控、几丁质分解代谢、蛋白质异源二聚化活性、转录辅阻遏
蛋白活性、转录共调节因子活性、钙离子结合、对刺激的反应调控等过程在植物嫩芽的发育中都具有重要作用,直接或间接影响植物的生长、发育和逆境响应。叶绿体类囊体膜和光系统Ⅰ、光系统Ⅱ的发育对植物嫩芽的生长、发育和能量供给至关重要。
2.5 十里香茶树嫩芽差异基因KEGG功能分类注释
KEGG注释结果可以分为细胞过程、环境信息处理、遗传信息处理、代谢、有机系统和组织系统6类,共注释1 335条通路,其中代谢富集到的基因数量最多,为777条,其次为遗传信息处理,为378条,环境信息处理、有机系统、细胞过程、组织系统分别富集到81、43、36、20条(图5)。结果表明,在十里香茶树嫩芽发育阶段,植物的代谢和遗传信息处理的生物活动占据着重要的位置。从KEGG富集结果选取各cluster最显著的KEGG通路富集项(表3)。结果表明,13个cluster中差异基因表达主要涉及氨基酸生物合成、氨基糖
和核苷酸糖代谢等通路,均与蛋白质代谢过程紧密相关。蛋白质是生命活动的基因组成部分,植物生长发育过程中的所有关键步骤都与蛋白质表达密切相关,如光合作用、呼吸作用、营养物质吸收和转化、逆境响应和信号传递等。这些差异基因表达的富集结果为茶树发育过程中蛋白质代谢提供了重要线索,有助于更全面地理解十里香茶树嫩芽在生长发育阶段的生物学调控机制。
3 讨论
本研究应用空间转录组测序技术识别十里香茶树嫩芽的基因表达情况和空间信息。Spot聚类注释嫩芽不同细胞类型,划分13个不同细胞类型cluster,空间转录组图谱表明,不同细胞类型cluster在嫩芽不同发育时期的表达位置存在空间异质性。推测嫩芽作为茶树幼嫩组织,处于高度发育状态,细胞处于不断分裂分化过程,导致一些基因在特定阶段的特定细胞类型中表达,执行特定的生物学功能。
进一步鉴定不同细胞类型cluster的差异基因,发现LOC114312694、LOC114319171、LOC114320792、LOC114287723、LOC114284011、LOC114289235基因参与嫩芽的抗逆性调节,在嫩芽的幼叶部位高表达,在芽轴低表达。这可能是因为在茶树的嫩芽中,幼叶包裹着芽轴,叶片充当了芽轴的保护外层,芽轴受外界的影响远小于嫩叶,所以抗逆性基因在芽轴的表达较少而在幼叶中表达量较高,是机体适应外界环境机制的体现。LOC114263486、LOC114320821、LOC114292779、LOC114321117、LOC114286858基因参与生长发育调控,在幼叶中高表达,在芽轴中低表达。这可能是由于幼叶比芽轴接触到更多的光,光合作用更强,因此这些与生长发育调控相关的基因在幼叶中高表达。LOC1142266906是类黄酮代谢途径下游的无色花青素还原酶(LAR),LOC1142266906基因在整个嫩芽组织中高表达,说明类黄酮代谢在十里香茶树嫩芽发育中发挥重要的作用。
基于空间转录组图谱分析嫩芽中特定细胞类型的表达模式,鉴定了大量在十里香茶树嫩芽不同部位间表达水平显著差异的基因,且涉及多个生物学过程和功能。GO功能富集分析结果显示,细胞组分中主要富集于细胞部分、细胞器等;在分子功能中主要富集于结合、蛋白和酶调节活性部分;显著的GO富集通路包括翻译、核糖体、核小体、细胞壁大分子分解代谢、转录辅阻遏蛋白活性、转录共调节因子活性、钙离子结合、对刺激的反应调控、茉莉酸介导信号通路调控、几丁质分解代谢、蛋白质异源二聚化活性、叶绿体类囊体膜和光系统Ⅰ、光系统Ⅱ等,这些通路直接或间接影响着嫩芽的生长发育、能量供给和逆境胁迫反应,在嫩芽发育过程中具有重要作用。KEGG数据库共注释到1 335条代谢通路,代谢占据主导位置,与Muthusamy等[30]研究结果相似,基因富集的代谢途径包括氨基酸代谢、碳代谢等代谢调节,花青素、生长素、植物激素等前提物质的合成,植物信号传导,以及耐寒、耐旱、耐抗病等抗逆胁迫。综合富集分析发现,嫩芽基因调控网络主要参与逆境胁迫、生长发育等过程,反映了嫩芽对生态环境变化的适应性,在发育阶段需要平衡生长发育和应对外部逆境的需求,以确保其在不同生态条件下生长发育。
参考文献
49程琳, 郝艳林, 曹思睿, 等. 茶树基因组研究进展[J]. 信阳师范学院学报(自然科学版), 2021, 34(4): 606-613.
Cheng L, Hao Y L, Cao S R, et al. Advances in genome research of tea plant [J]. Journal of Xinyang Normal University (Natural Science Edition), 2021, 34(4): 606-613.
50Xia E H, Zhang H B, Sheng J. The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis [J]. Molecular Plant, 2017, 10(6): 866-877.
51Wei C L, Yang H, Wang S B, et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality [J]. PNAS, 2018, 115(18): E4151-E4158.
52Xia E H, Tong W, Wu Q, et al. Tea plant genomics: achievements, challenges and perspectives [J]. Horticulture Research, 2020, 7: 7. doi: 10.1038/s41438-019-0225-4.
53Xia E H, Tong W, Hou Y, et al. The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into its genome evolution and adaptation [J]. Molecular Plant, 2020, 13(7): 1013-1026.
54Chen J D, Zheng C, Ma J Q, et al. The chromosome-scale genome reveals the evolution and diversification after the recent tetraploidization event in tea plant [J]. Horticulture Research, 2020, 7: 63. doi: 10.1038/s41438-020-0288-2.
55Wang X C, Feng H, Chang Y X, et al. Population sequencing enhances understanding of tea plant evolution [J]. Nature Communications, 2020, 11: 4447. doi: 10.1038/s41467-020-
18228-8.
56Lin P, Wang K L, Wang Y P, et al. The genome of oil-camellia and population genomics analysis provide insights into seed oil domestication [J]. Genome Biology, 2022, 23: 14. doi: 10.1186/s13059-021-02599-2.
57Xiang P, Zhu Q F, Zhang L H, et al. Integrative analyses of transcriptome and metabolome reveal comprehensive mechanisms of epigallocatechin-3-gallate (EGCG) biosynthesis in response to ecological factors in tea plant (Camellia sinensis) [J]. Food Research International, 2023, 166: 112591. doi: 10.1016/j.foodres.2023.112591.
58Zhang Y R, Fu J M, Zhou Q Y, et al. Metabolite profiling and transcriptome analysis revealed the conserved transcriptional regulation mechanism of caffeine biosynthesis in tea and coffee plants [J]. Journal of Agricultural and Food Chemistry, 2022, 70(10): 3239-3251.
59Zheng Y C, Wang P J, Chen X J, et al. Integrated transcriptomics and metabolomics provide novel insight into changes in specialized metabolites in an albino tea cultivar (Camellia sinensis (L.) O. Kuntz) [J]. Plant Physiology and Biochemistry, 2021, 160: 27-36.
60Chang L, Jing L, Li Y L, et al. A spatiotemporal atlas of organogenesis in the development of orchid flower [J]. Nucleic Acids Research, 2022, 50(17): 9724-9737.
61李方东. 茶树转录组组装评估与多组学生物信息平台开发[D]. 合肥: 安徽农业大学, 2021.
Li D F. Evaluation of tea transcriptome assembly and development of multi-group student information platform [D]. Hefei: Anhui Agricultural University, 2021.
62刘芳芳, 刘文祥, 郑伟, 等. 茶树NF-Y基因家族鉴定及非生物胁迫下的表达分析[J]. 江苏农业科学, 2023, 51(5): 81-93.
Li F F, Liu W X, Zheng W, et al. Identification of NF-Y gene family in tea tree and expression analysis under abiotic stresses [J]. Jiangsu Agricultural Sciences, 2023, 51(5): 81-93.
63Shi C Y, Yang H, Wei C L, et al. Deep sequencing of the Camellia sinensis transcriotome revealed candidate genes for major metabolic pathways of tea-specific compounds [J]. BMC Genomics, 2011, 12: 131. doi: 10.1186/1471-2164-12-
131.
64Qiu H J, Zhang X L, Zhang Y J, et al. Depicting the genetic and metabolic panorama of chemical diversity in the tea plant [J]. Plant Biotechnology Journal, 2024, 22(4): 1001-1016.
65Lei X G, Li H Y, Li P P, et al. Genome-wide association studies of biluochun tea plant populations in Dongting mountain and comprehensive identification of candidate genes associated with core agronomic traits by four analysis models [J]. Plants, 2023, 12(21): 3179. doi: 10.3390/plants12213719.
66Wang X C, Zhao Q Y, Ma C L, et al. Global transcriptome profiles of Camellia sinensis during cold acclimation [J]. BMC Genomics, 2013, 14: 415. doi: 10.1186/1471-2164-14-415.
67Qiu H J, Zhu X, Wan H L, et al. Parallel metabolomic and transcriptomic analysis reveals key factors for quality improvement of tea plants [J]. Journal of Agricultural and Food Chemistry, 2020, 68(19): 5483-5495.
68Giacomello S, Salmén F, Terebieniec B K, et al. Spatially resolved transcriptome profiling in model plant species [J]. Nature Plants, 2017, 3: 17061. doi: 10.1038/nplants.2017.61.
69Moreno-Villena J J, Zhou H R, Gilman I S, et al. Spatial resolution of an integrated C4+CAM photosynthetic metabolism [J]. Science Advances, 2022, 8(31): 2349. doi: 10.1126/sciadv.abn2349.
70Song X H, Guo P R, Xia K K, et al. Spatial transcriptomics reveals light-induced chlorenchyma cells involved in promoting shoot regeneration in tomato callus [J]. PNAS, 2023, 120(38): e2310163120. doi: 10.1073/pnas.2310163120.
71Yang X L, Poelmans W, Grones C, et al. Spatial transcriptomics of a lycophyte root sheds light on root evolution [J]. Current Biology, 2023, 33(19): 4069-4084.
72王玮, 刘娜, 徐亚文, 等. 澜沧江中下游流域古茶树资源儿茶素含量的多样性分析[J/OL]. 分子植物育种, 2022: 1-17[2023-09-12]. http://kns.cnki.net/kcms/detail/46.1068.
S.20220720.1902.009.html.
Wang W, Liu N, Xu Y W, et al. Diversity analysis of catechin content of ancient tea tree resources in the middle and lower reaches of Lancang River [J/OL]. Molecular Plant Breeding, 2022: 1-17[2023-09-12]. http://kns.cnki.net/ kcms/
detail/46.1068.S.20220720.1902.009.html.
73沈雪梅, 杨丕琼, 许文徽, 等. 十里香茶产业化发展的都市农庄模式[J]. 云南农业, 2014(11): 42-43.
Shen X M, Yang P Q, Xu W H, et al. Urban farm model of Industrialization development of Shilixiang tea [J]. Yunnan Agriculture, 2014(11): 42-43.
74沈晓进. 昆明十里香古茶树保护与利用的探讨[J]. 西南林学院学报, 2004(2): 27-29.
Shen X J. Discussion on protection and utilization of Kunming Shilixiang ancient tea tree [J]. Journal of Southwest Forestry College, 2004(2): 27-29.
75沈雪梅, 杨丕琼, 许文徽, 等. 云南十里香茶的保护现状及发展研究[C]//中国科学技术协会, 云南省人民政府. 第十六届中国科协年会——分12茶学青年科学家论坛论文集. 昆明: [出版者不详], 2014: 40-43.
Shen X M, Yang P Q, Xu W H, et al. Study on conservation status and development of Shilixiang tea in Yunnan [C]//China Association for Science and Technology, People's Government of Yunnan Province. The 16th Annual Meeting of China Association for Science and Technology: 12 Collection of Essays for the Forum of Young Tea Science Scientists. Kunming: [s.n.], 2014: 40-43.
76McInnes L, Healy J, Melville J. UMAP: uniform manifold approximation and projection for dimension reduction [J]. ArXiv, 2018: 1802.03426. doi: 10.48550/arXiv.1802.03426.
77Van der Maaten L. Accelerating t-SNE using tree-based algorithms [J]. Journal of Machine Learning Research, 2014(15): 3221-3245.
78Muthusamy M, Kim J Y, Yoon E K, et al. BrEXLB1, a Brassica rapa expansin-like B1 gene is associated with root development, drought stress response, and seed germination [J]. Genes, 2020, 11(4): 404. doi: 10.3390/genes11040404.
猜你喜欢
动物医学进展(2024年4期)2024-04-10
世界中医药(2021年17期)2021-09-28
广西植物(2020年9期)2020-11-02
中国现代医生(2020年12期)2020-07-04
心电与循环(2020年1期)2020-02-27
风湿病与关节炎(2019年10期)2019-12-02
山东农业科学(2019年7期)2019-09-03
风湿病与关节炎(2019年2期)2019-04-15
中国中药杂志(2017年15期)2017-08-30
江苏农业科学(2017年5期)2017-04-15
