
茶树橙花叔醇和芳樟醇樱草糖苷含量全基因组关联分析及候选基因预测
2024-03-28 02:41张力岚王让剑
作物学报 2024年4期
张力岚 杨 军 王让剑,*
茶树橙花叔醇和芳樟醇樱草糖苷含量全基因组关联分析及候选基因预测
张力岚1,2杨 军1,2王让剑1,2,*
1福建省农业科学院茶叶研究所, 福建福州 350013;2国家茶树改良中心福建分中心, 福建福州 350013
橙花叔醇与芳樟醇是广泛分布于植物中的挥发性萜烯醇类化合物, 在茶树新梢中主要以樱草糖苷形式存在, 提高其含量对茶叶香气品质改良具有重要意义。为揭示茶树橙花叔醇和芳樟醇樱草糖苷的遗传机制, 本研究以169个茶树自然杂交后代单株为关联群体, 利用均匀分布在茶树染色体上的675,245个SNP标记, 对3个年份下茶树新梢中橙花叔醇与芳樟醇樱草糖苷含量进行全基因组关联分析(genome-wide association study, GWAS)。结果表明, 橙花叔醇与芳樟醇樱草糖苷含量的表型变异系数为60.83%~80.08%, 广义遗传力分别为51.29%与61.87%, 基本符合正态分布, 具有典型的数量性状遗传特征。全基因组关联分析共检测到50个显著关联位点, 各位点分别对橙花叔醇和芳樟醇樱草糖苷含量变化的贡献率均超过20%, 其中橙花叔醇樱草糖苷含量(nerolidol primeveroside content, NPC)变化位点最大贡献率为38.73%, 芳樟醇樱草糖苷含量(linalool primeveroside content, LPC)变化位点最大贡献率为39.07%。通过等位变异效应分析, 鉴定出4个主效SNP位点及其优异等位变异, 并发现1个可同时调控NPC和LPC的一因多效位点。结合已有的文献报道和基因的功能注释, 筛选出每个显著位点置信区间内最有可能的候选基因共59个, 主要涉及茶树的糖类代谢、转录调控、萜烯类生物合成等多个生物学过程, 其中26个基因在适制绿茶品种和适制乌龙茶品种单芽中的表达水平存在显著差异。本研究为深入解析茶树橙花叔醇和芳樟醇樱草糖苷含量的遗传机制提供了新的信息, 为加速培育高香型优异茶树新品种提供了重要的基因资源。
茶树; 橙花叔醇樱草糖苷; 芳樟醇樱草糖苷; 全基因组关联分析; 候选基因
茶树是世界上最重要的经济作物之一, 2021年在全球的种植面积约为525万公顷, 产量达到2819万吨(http://faostat.fao.org/)。香气是茶树重要的感官品质之一, 很大程度影响茶叶的市场价值和大众消费者的购买欲望。因此, 香气品质的优劣一直都是茶树育种工作者最关注的性状之一。阐明茶树香气品质相关性状的合成调控机理, 对提高茶树育种的效率和准确性、推动茶产业高质量发展具有重要的研究意义。
橙花叔醇属于倍半萜烯醇, 有水果百合、花木香和木香的香韵。芳樟醇又名沉香醇, 属单萜烯醇类, 有玉兰或百合花的香气。橙花叔醇与芳樟醇在茶树体内主要以樱草糖苷(二糖糖苷)的形式存在, 且二者均为茶叶中极具代表性的香气成分, 对茶叶香气的形成有重要的作用[1]。Liu等[2]通过揭示茶树中橙花叔醇与芳樟醇的生物合成调控机制, 为改善茶叶香气品质提供了新的研究思路。Wei等[3]基于高通量SNP芯片, 定位到一个与绿茶特征香气成分(E)-异丁香酚生物合成相关的强选择位点。茶叶中关键呈香成分生物合成及其调控机制的研究报道, 为茶叶香气品质的提升提供了重要的研究基础和科学依据。近年来, 利用全基因组关联分析(genome wide association analysis, GWAS)技术进行位点和基因挖掘已经广泛应用在各类植物的香气性状研究中。在桃树中, 对256份种质资源成熟果实中的挥发性化合物进行了测定, 基于全基因组关联分析定位到一个编码萜烯合成酶的基因, 该基因位于已报道的数量性状基因座(quantitative trait locus, QTL)区域内, 且与芳樟醇的生物合成有关[4]。在西瓜中, 利用包括GWAS在内的多组学技术, 在4号染色体上挖掘到一个类胡萝卜素裂解双加氧酶(carotenoid cleavage dioxygenase, CCD)基因, 该基因已被证实参与萜类挥发性有机物(volatile organic compounds, VOCs)的合成与代谢[5]。在蓝莓中, 研究人员对886份个体的17种VOCs进行了测定, 通过GWAS关联到6个与芳樟醇有关的基因, 8个与桉叶醇有关的基因[6]。
茶树性状遗传背景十分复杂, 与香气品质相关的基础研究一直是茶树遗传育种的难点和重点。迄今, 有关茶树橙花叔醇樱草糖苷含量(nerolidol primeveroside content, NPC)和芳樟醇樱草糖苷含量(linalool primeveroside content, LPC)全基因组关联分析的研究报道较少, 遗传机制尚不明晰。已经定位克隆的位点或基因, 并不能完全解释茶树香气品质相关性状的遗传变异, 其复杂的分子机制尚需进一步研究。
本研究以169个不同遗传背景的茶树自然杂交后代单株作为关联群体, 结合包含675,245个SNP标记的简化基因组测序数据, 对NPC和LPC性状展开全基因组关联分析, 挖掘茶树群体中调控NPC和LPC的重要位点及候选基因, 为解析茶树橙花叔醇和芳樟醇樱草糖苷含量的遗传机制, 推进高香型茶树新品种的选育提供参考。
1 材料与方法
1.1 试验材料
供试材料为包含169个茶树种质的关联群体, 为福建省农业科学院茶叶研究所构建的高香型优异茶树种质FT516、FT104的自然杂交后代单株, 保存于福建省乌龙茶种质资源圃内(27°13′40″N, 119°32′11″E) (附表1)。其中FT516为黄观音(♀)×铁观音(♂)杂交后代, FT104为毛蟹(♀)自然杂交后代。沙培从母树上获得的成熟种子, 2015年3月单株种植于田间, 株距0.5 m, 行距1.5 m, 每份种质材料为单株种植, 田间管理措施相对一致。取相同生长环境下的已推广品种‘福云6号’与‘悦茗香’的健康无损伤单芽, 通过转录组测序, 辅助筛选候选基因。其中, ‘福云6号’为适制绿茶品种, 新梢单芽中NPC和LPC较低, ‘悦茗香’为适制乌龙茶的高香型品种, 新梢单芽中NPC和LPC较高。
1.2 表型数据测定与分析
2018、2019和2020连续3年分别采摘群体内各种质春季新梢健康无损伤单芽, 每份材料确保有3个生物学重复, 用于测定橙花叔醇樱草糖苷与芳樟醇樱草糖苷的含量。
称取2 g嫩芽置于50 mL塑料离心管中(精确至0.01 g), 加入20 mL甲醇, 10,000转 min-1均质2 min, 超声20 min, 1960´离心5 min, 移取上清液过0.22 µm有机滤膜进样。
色谱条件: 流速0.3 mL min–1; 柱温40℃; 样品盘温度15℃; 进样量2 μL; 流动相A为0.1% (v/v)甲酸水溶液, 流动相B为乙腈; 梯度洗脱条件为: 0~3 min, 10%B; 3~9 min, 25%B; 9~11 min, 40%B; 11~12 min, 90%B; 12.0~12.1 min, 90%B; 12.1~ 16.0 min, 10%B。
质谱条件: 离子源为电喷雾离子源ESI; 负离子模式; 检测方式为多反应监测; 大气压电离(API)气体为氮气(纯度>95%, 压力600~900 kPa); 碰撞气体为高纯氩; 界面电压3.0 kV; 界面温度300℃; 脱溶剂温度250℃; 干燥气10 L min–1, 雾化气3 L min–1, 加热气10 L min–1, 热块温度400℃。
称取橙花叔醇樱草糖苷与芳樟醇樱草糖苷标准品各10 mg, 用甲醇溶解并定容至10 mL, 即为1 mg mL–1的单标储备液, –20℃下贮存于密封的棕色玻璃瓶中, 用其配制成不同浓度梯度的橙花叔醇樱草糖苷与芳樟醇樱草糖苷标准溶液待用。将标准溶液进样, 以标准溶液质量浓度为横坐标, 定量离子峰面积为纵坐标, 绘制标准曲线并分别对待测种质橙花叔醇樱草糖苷与芳樟醇樱草糖苷进行定量。
采用Microsoft Excel 2021, 对关联群体的表型数据进行整理。利用软件SPSS 26.0, 进行表型性状的统计描述以及广义遗传力(2)的计算[7]。采用皮尔逊(Pearson)相关分析, 计算不同年份间茶树新梢中各糖苷含量间的相关系数。
1.3 全基因组关联分析
经过III酶切茶树的基因组DNA、酶切片段3¢端加A处理、连接Dual-index测序接头、PCR扩增纯化以及目的片段文库质检等一系列主要流程, 对待测群体的种质资源进行SLAF-seq测序(specific locus amplified fragment sequencing)。以上工作基于北京百迈客生物科技有限公司的Illumina HiSeq测序平台完成。以舒茶早品种基因组序列[8]作参考, 对获得的10,802,426个SNP标记进行质控筛选, 根据最小等位基因频率(MAF)≥0.05, 缺失率(miss)≤0.2的标准进行过滤, 保留675,245个SNP标记用于后续分析[9]。
利用过滤质控后的高质量SNP标记, 对关联材料的群体结构(值)和亲缘关系(值)进行了分析。使用软件TASSEL 5.0的混合线性模型(mixed linear model, MLM), 以Q和K作协变量, 对橙花叔醇和芳樟醇樱草糖苷含量在单一年份下的表型值进行全基因组关联分析。显著性阈值设置为:<1.48×10–6[=(10)–1,为使用标记的总数], 因此–log10()≥6.0时, 认为该标记与目的性状显著关联。
1.4 等位变异分析
利用重要SNP位点对供试群体进行基因分型, 计算不同等位变异对应表型的均值, 单因素方差分析比较不同等位变异间糖苷含量的差异。利用数据分析平台SangerBox绘制箱线图, 展示位点的等位变异效应。
1.5 候选基因的筛选
以关联群体连锁不平衡的衰减距离(100 kb)为连锁区间, 基于数据库TeaGVD[10](http://www.teaplant. top/teagvd)、NCBI (https://www.ncbi.nlm.nih.gov/)以及舒茶早品种参考基因组[8]的注释信息, 在显著关联位点上下游各100 kb的范围内, 初步筛选候选基因。利用eggNOG-mapper (http://eggnog-mapper. embl.de/)数据库, 对关联基因的功能进行预测与注释, 辅助候选基因的筛选。基于TPIA[8](http://tpia. teaplants.cn/)数据库, 检索候选基因在茶树8个代表性组织(顶芽、花、果、嫩叶、成熟叶、老叶、根、茎)中的表达数据。取已推广的栽培品种‘福云6号’与‘悦茗香’的春梢单芽作为样品, 在北京百迈客生物科技有限公司(Biomarker Technologies)进行转录组测序, 每个样品确保3个生物学重复。利用百迈客云分析平台(https://international.biocloud.net/)完成2个品种间差异表达基因(differentially expressed genes, DEGs)的筛选, 将已获得的GWAS候选基因在DEGs中搜索, 最终得到基因表达存在差异的GWAS候选基因。
2 结果与分析
2.1 表型描述统计分析
对3个年份下(E2018、E2019及E2020)共169份材料NPC与LPC的表型值进行统计分析发现, 该群体材料的NPC和LPC在不同年份间均存在明显的差异(图1)。

图1 关联群体在不同年份下表型值的箱线图
对于NPC, 3个年份下的均值在0.0049~0.0144 µg mL–1之间, 表型变异范围在0.0013~0.0508 µg mL–1之间, 变异系数在72.55%~80.08%; 对于LPC, 3个年份下的均值在0.0073~0.0139 µg mL–1之间, 表型变异范围在0.0014~0.0500 µg mL–1之间, 变异系数在60.83%~66.71% (表1)。NPC和LPC在3个年份下的变异系数均大于60%, 说明该关联群体在2个目标性状中存在丰富的表型变异。NPC与LPC的峰度和偏度值在0.77~2.81之间, 均小于3, 近似正态分布, 属于较典型的数量性状, 符合关联分析的基本要求。相关性分析结果表明, NPC与LPC在不同年份间均存在极显著的正相关(<0.01), NPC的相关系数在0.58~0.76之间, LPC的相关系数在0.59~0.78之间(图2)。计算NPC和LPC的广义遗传力分别为51.29%和61.87% (表1)。以上结果表明, 这2个性状的遗传效应主要受基因控制, 但在一定程度上也会受到环境因素的影响。
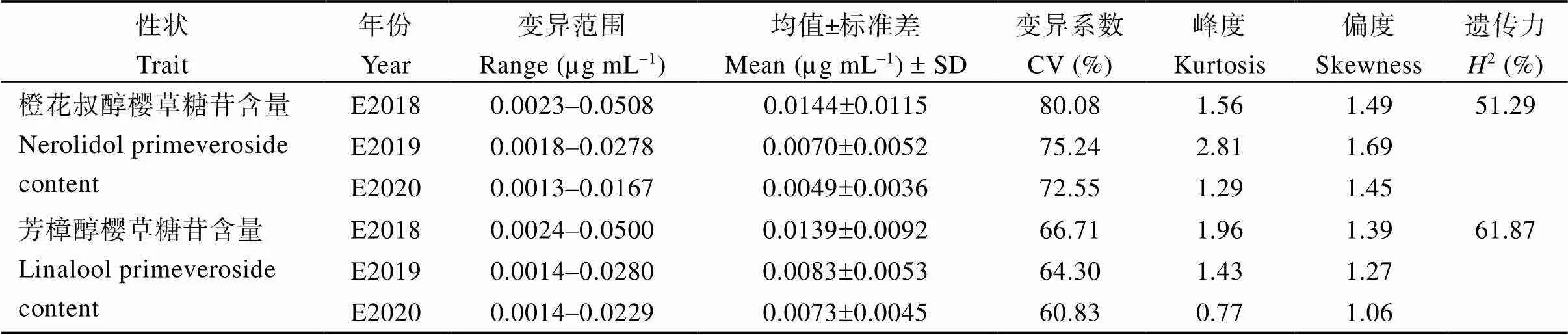
表1 不同年份间橙花叔醇与芳樟醇樱草糖苷含量的描述统计分析
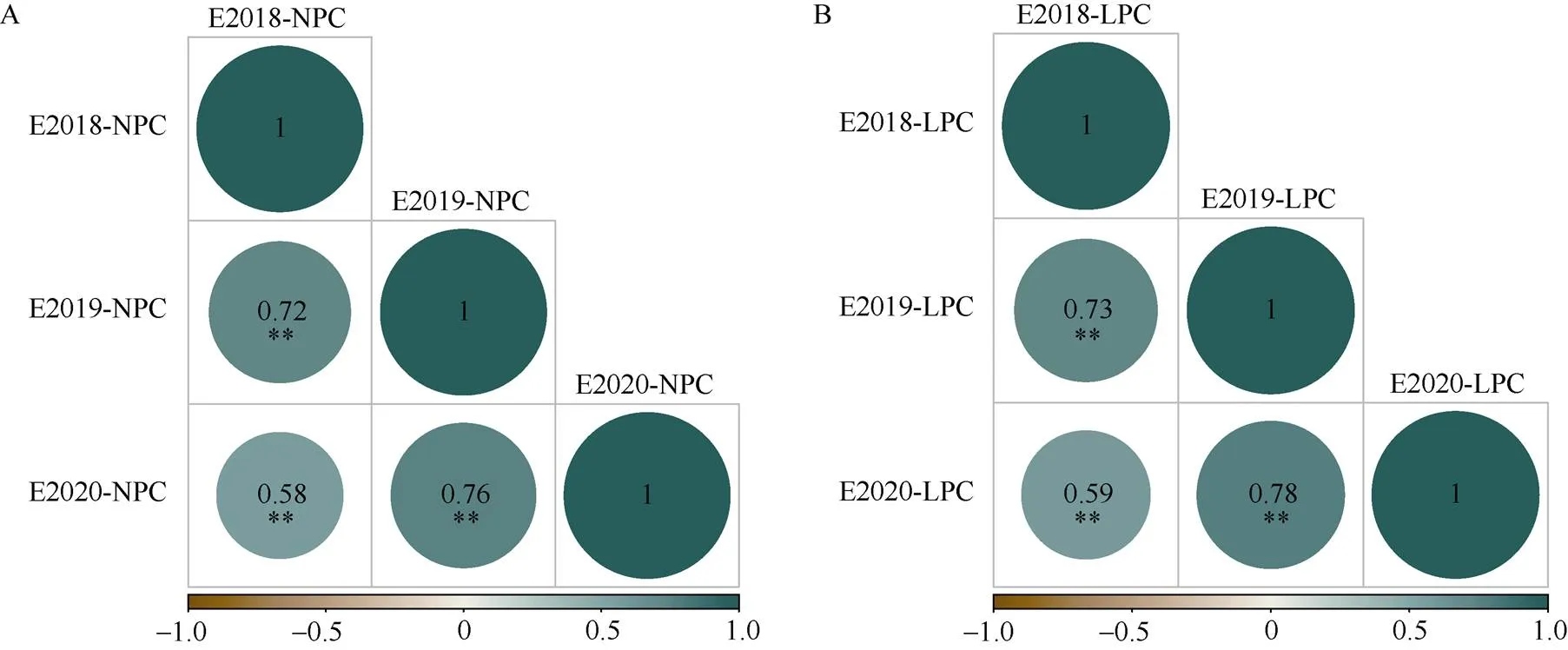
图2 不同年份间橙花叔醇与芳樟醇樱草糖苷含量的相关性分析
A: 橙花叔醇樱草糖苷含量(NPC); B: 芳樟醇樱草糖苷含量(LPC)。**表示在0.01概率水平差异显著。
A: nerolidol primeveroside content (NPC); B: linalool primeveroside content (LPC).** represents significant difference at the 0.01 probability level.
2.2 全基因组关联分析
本研究基于混合线性模型, 利用675,245个SNP标记对NPC和LPC进行全基因组关联分析, 筛选到显著SNP位点共50个(表2)。在6次独立的关联事件中,共有28个位点被检测到2次及其以上, 4个位点(SNP_2、SNP_3、SNP_39和SNP_40)被检测到3次, 可将其视为稳定的高可信度标记位点, 可能含有控制茶树NPC和LPC的保守基因。其中, NPC共检测到39个显著SNP, 贡献率在21.06%~38.73%之间,值最小的位点为SNP_2, 在E2018检测到的SNP位点有4个, E2019与E2020分别为23个和35个, 有21个被检测到2次及其以上, 2个被检测到3次。LPC共检测到12个显著SNP, 贡献率在24.98%~39.07%之间,值最低的位点为SNP_40, 在E2018检测到的SNP位点有3个, E2019与E2020分别为7个和10个, 有7个被检测到2次及其以上, 1个被检测到3次(表2)。SNP_39 (Sca.1970:1955068)与NPC和LPC均为显著关联, 说明该位点附近可能存在一因多效。

表2 橙花叔醇与芳樟醇樱草糖苷含量显著关联的SNP位点
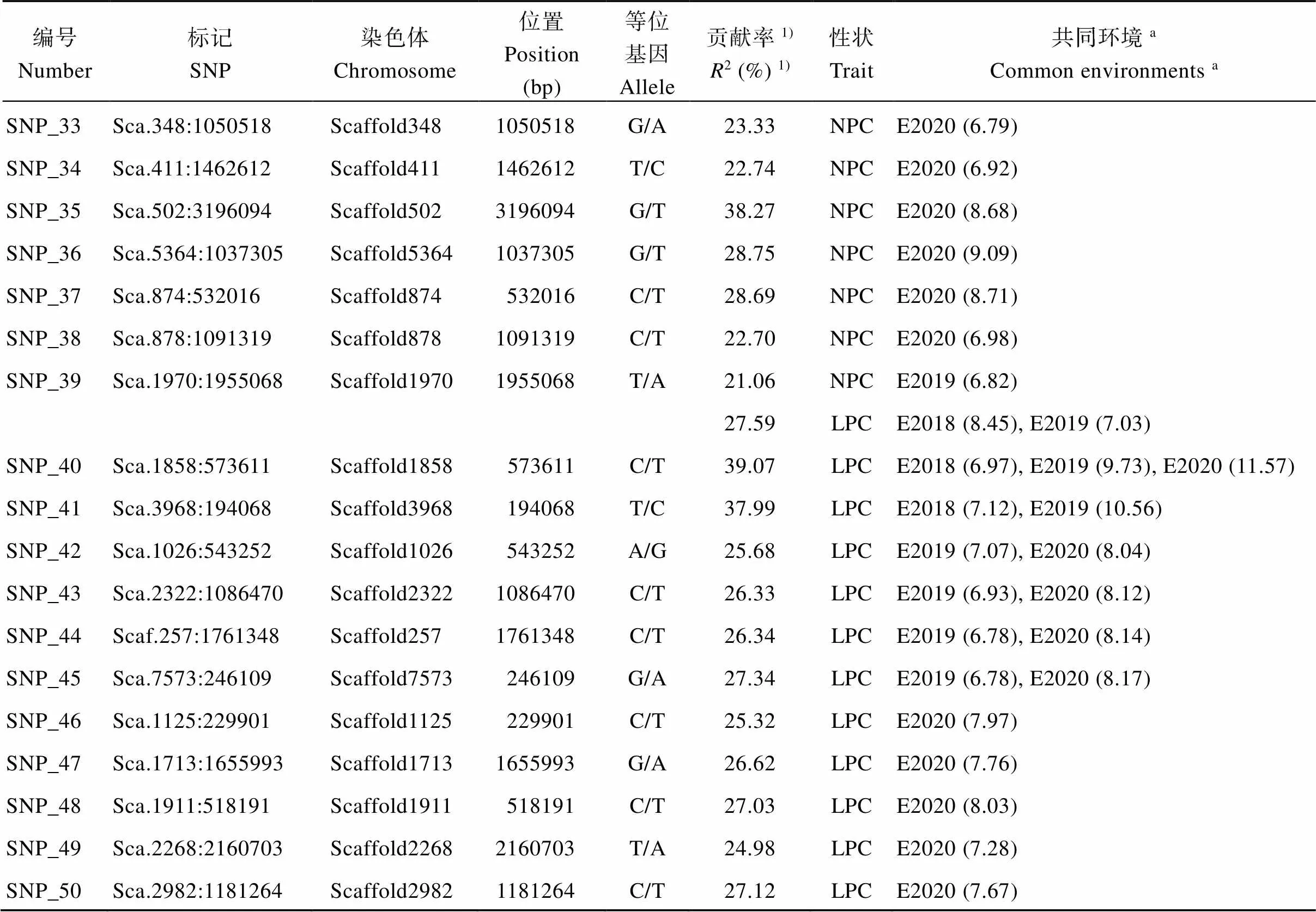
(续表2)
a: 括号中的数字代表在每个鉴定年份中检测到的SNP的–log10()值;1):2为在多个年份间检测的最高值。NPC: 橙花叔醇樱草糖苷含量; LPC: 芳樟醇樱草糖苷含量。
a: the numbers in parentheses represent the –log10() values of SNP detected in each test years;1): only the highest value of2detected in multiple years was showed. NPC: nerolidol primeveroside content; LPC: linalool primeveroside content.
2.3 重要位点等位变异鉴定及效应分析
结合表型数据分析发现, NPC和LPC共有4个主效SNP位点的等位变异在3年间均可导致其表型差异呈极显著水平(图3)。位点SNP_2 (Sca.2168:1897613)的基因型CC可使NPC提高0.0031~0.0068 µg mL–1, 因此CC为优异等位变异; 位点SNP_3 (Sca.2542:459511)的基因型CC可使NPC提高0.0003~0.0126 µg mL–1, 因此CC为优异等位变异; 位点SNP_49 (Sca.2268: 2160703)的基因型TT可使LPC提高0.0031~0.0070 µg mL–1, 因此TT为优异等位变异。

(图3)
NPC: 橙花叔醇樱草糖苷含量; LPC: 芳樟醇樱草糖苷含量。NPC: nerolidol primeveroside content; LPC: linalool primeveroside content.
此外, 与NPC和LPC共同关联的位点SNP_39 (Sca.1970:1955068)在2个性状中的有利等位基因是相同的, 均为AA。该等位变异可导致E2018、E2019和E2020年份的平均NPC比等位变异TT分别提高0.0086、0.0068和0.0072 µg mL–1, LPC则分别提高0.0102、0.0072和0.0074 µg mL–1, 其差异均呈极显著水平(图3)。该主效SNP位点对茶树NPC和LPC均具备显著的调控效应, 其周围区域可能存在对茶树NPC和LPC具有重要调控作用的基因, 是茶树香气品质遗传改良的潜在重要区域。
2.4 候选基因的挖掘
以LD衰减距离100 kb[9]作为SNP位点上下游的候选区间, 基于茶树基因组的物理位置, 在舒茶早基因组上对50个候选区间进行基因扫描。根据SNP位点注释信息、基因表达水平以及相关研究报道, 最终筛选出59个与NPC和LPC相关的候选基因, 其中47个与NPC的变化有关, 13个与LPC的变化有关(附图1)。这些基因主要涉及编码萜烯合成、糖基化以及糖苷水解等多种类型, 其中还包括了一些重要的转录因子, 如WRKY、MYB、NAC。
在5个编码萜烯类化合物生物合成的基因中,编码ent-柯巴基焦磷酸合酶(ent-copalyl diphosphate synthase, CPS)蛋白,与编码α-萜品醇合酶蛋白,编码萜烯合成酶(terpene synthase, TPS)蛋白, 而编码的1-脱氧-D-木酮糖-5-磷酸合成酶(1-deoxy-D-xylulose 5-phosphate synthase, DXS)是萜烯合成2-C-甲基-D-赤藻糖醇-4-磷酸(2-C-Methyl- D-erythritol-4-phosphate, MEP)途径的关键酶之一, 其中与在顶芽与嫩叶中的表达量较高(附图1)。
在4个主效SNP位点中, SNP_49关联区域的候选基因为编码糖基转移酶的, SNP_2的候选基因为编码β-甘露糖苷酶的, 而SNP_3上的2个候选基因与均编码细胞色素P450 (cytochrome P450, CYP450)蛋白。其中,虽在不同茶树组织中均有表达, 但在老叶与成熟叶片中的表达量更高, 而与的表达水平则在顶芽与嫩叶中较高(附图1)。基因位于与NPC和LPC同时关联的标记SNP_39 (Sca.1970:1955068)的候选区间上, 编码BAHD酰基转移酶蛋白, 只在幼嫩叶片组织中特异性表达(附图1), 参与植物次生代谢产物如挥发性芳香化合物合成修饰的重要过程[11]。
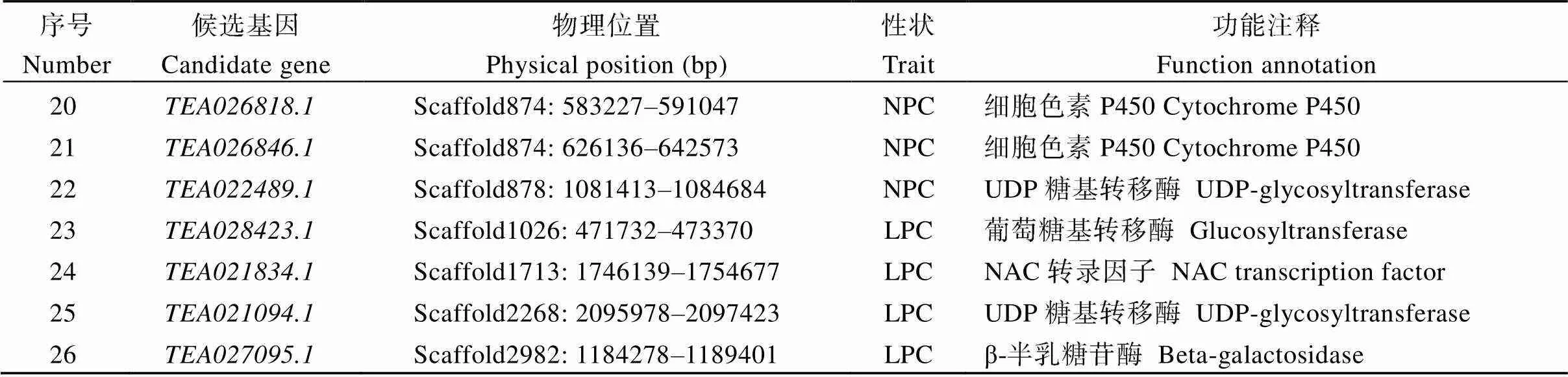
进一步分析发现, 59个GWAS候选基因中有26个基因的表达水平在绿茶品种‘福云6号’与乌龙茶品种‘悦茗香’的新梢单芽中存在显著差异(表3和图4)。其中, 8个候选基因编码催化萜类生物合成的CYP450活性蛋白, 分别为、、、、、、和; 3个编码糖苷水解酶活性蛋白, 分别为、和; 12个编码糖基转移酶活性蛋白, 分别为、、、、、、、、、、和; 2个WRKY转录因子和, 以及1个NAC转录因子。
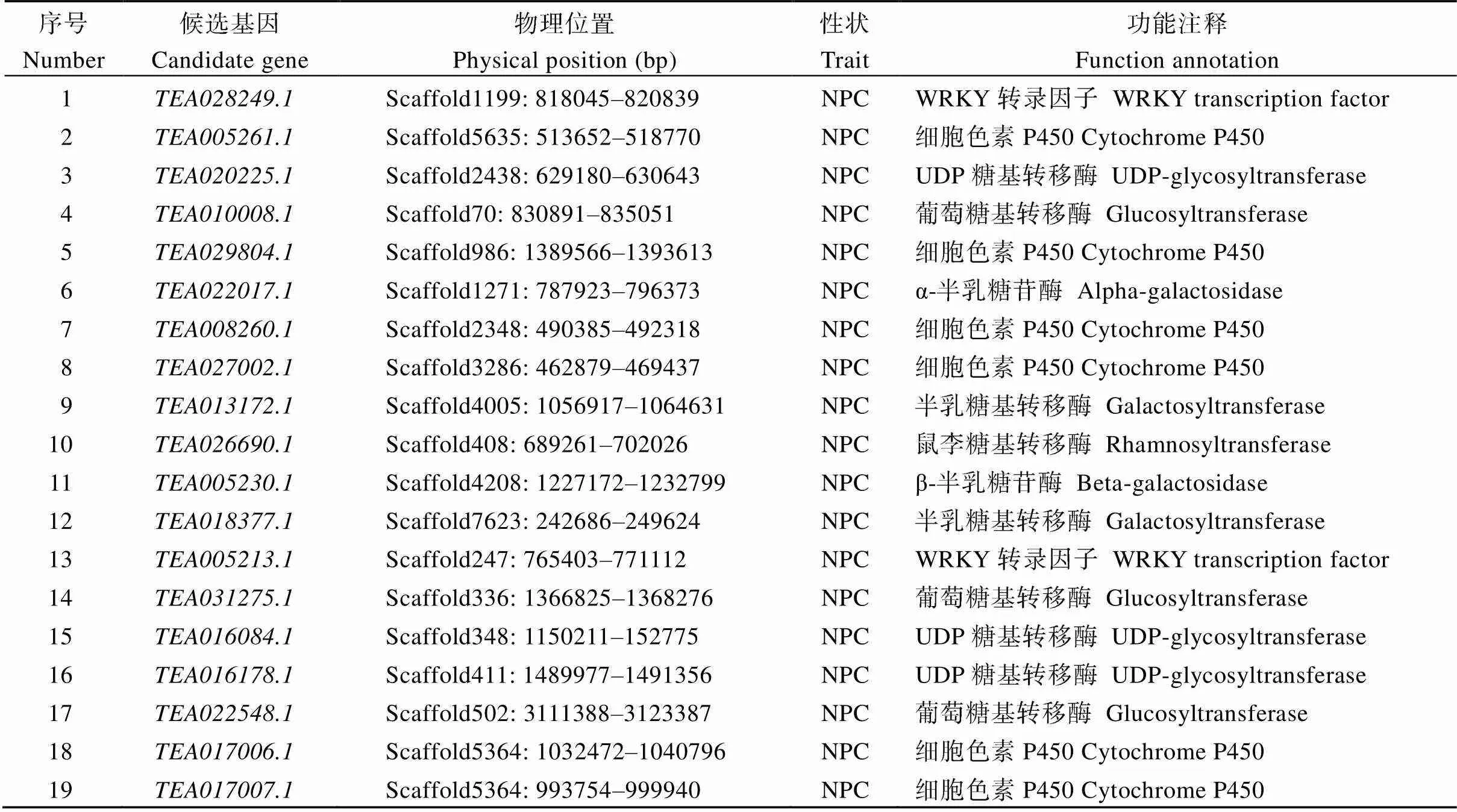
表3 候选基因鉴定
(续表3)
NPC: 橙花叔醇樱草糖苷含量; LPC: 芳樟醇樱草糖苷含量。NPC: nerolidol primeveroside content; LPC: linalool primeveroside content.
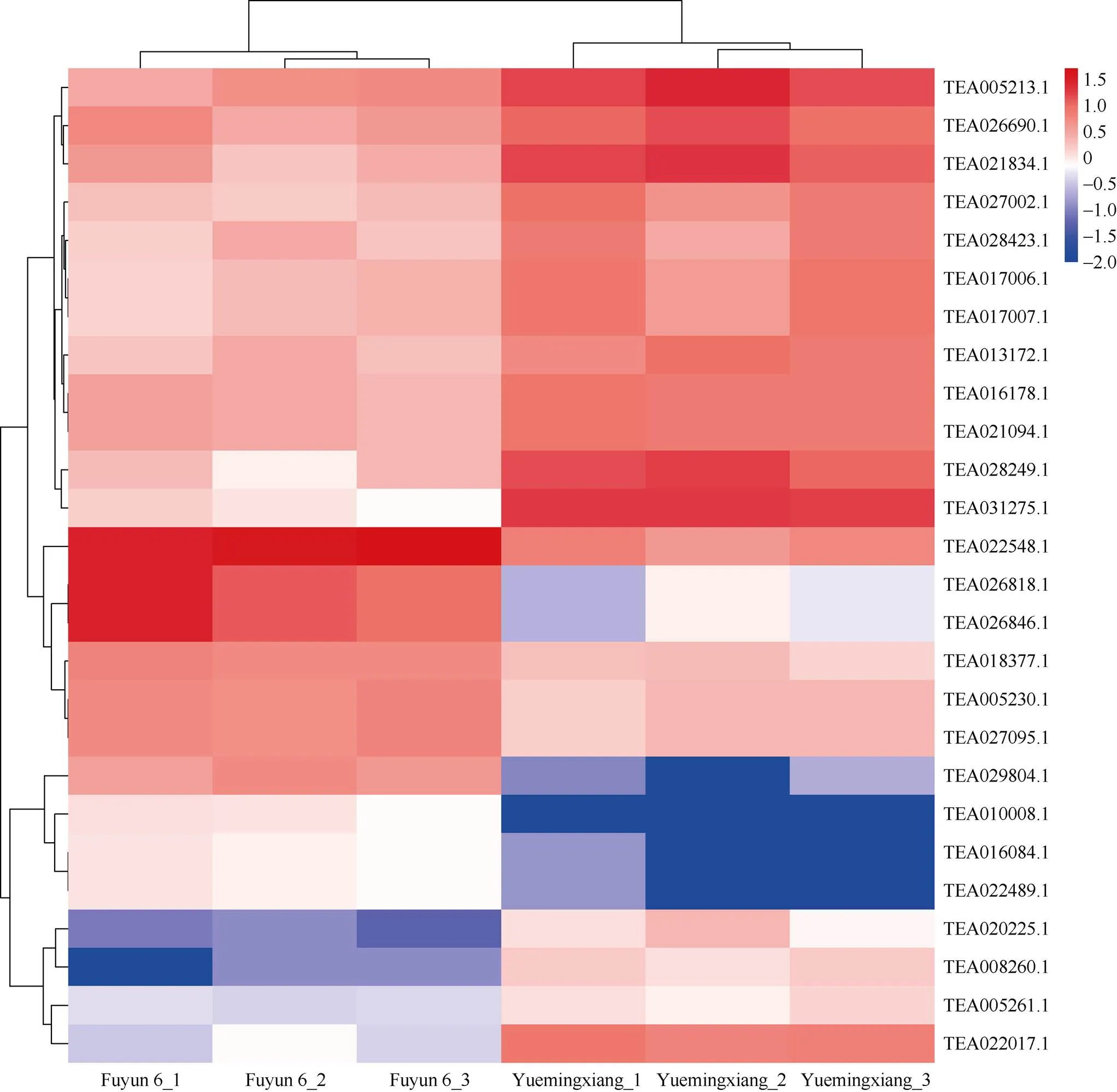
图4 转录组与GWAS筛选共有基因的表达水平热图
Fuyun 6_1、Fuyun 6_2、Fuyun 6_3: 候选基因在绿茶品种‘福云6号’单芽中的表达水平; Yuemingxiang_1、Yuemingxiang_2、Yuemingxiang_3: 候选基因在乌龙茶品种‘悦茗香’单芽中的表达水平。
Fuyun 6_1, Fuyun 6_2, Fuyun 6_3: expression level of candidate genes in buds of green tea variety ‘Fuyun 6’; Yuemingxiang_1, Yuemingxiang_2, Yuemingxiang_3: the relative expression level of candidate genes in buds of oolong tea variety ‘Yuemingxiang’.
这26个基因与茶树新梢中橙花叔醇与芳樟醇樱草糖苷合成积累相关, 并且在不同适制性茶树品种之间显著差异表达, 进一步说明这些基因对不同适制性茶树品种香气品质的形成具有重要作用, 可初步视为茶树橙花叔醇与芳樟醇樱草糖苷合成积累的候选基因。基于候选基因注释结果与前人的研究进展, 绘制出茶树橙花叔醇与芳樟醇樱草糖苷合成积累的调控模型示意图(图5)。
TFs: 转录因子; TPS: 萜烯合成酶; CYP450: 细胞色素P450; GHs: 糖苷水解酶; GTs: 糖基转移酶。
TFs: transcription factors; TPS: terpene synthase; CYP450: cytochrome P450; GHs: glycoside hydrolases; GTs: glycosyltransferases.
3 讨论
3.1 香气品质性状遗传特征分析
本研究结果显示, 3个年份中169份参试材料NPC和LPC性状的平均值基本符合正态分布, 这与之前Fang等[12]、Hazra等[13]、Huang等[14]以及Yamashita等[15]对其他品质性状的研究结果一致。基于相关性分析, 发现NPC与LPC在不同年份间均存在极显著的正相关(<0.01), 说明目标性状主要受遗传因素影响, 遗传改良前景可观。2018年NPC和LPC均显著高于其他年份, 但显著关联位点较少。
通过对表型性状的统计及其全基因组关联分析,发现2018年NPC和LPC均显著高于其他两年, 但关联到的显著位点却较少。这可能是因为, 2018年春季实验材料取样点所在茶区遭受严重晚霜冻害, 促使茶树体内NPC和LPC大量积累, 进而起到抵御寒冷的作用, 而2019年与2020年NPC和LPC表型值的区分度更好, 故检测到的显著位点更多。通过比较SNP在茶树基因组上的物理位置, 可发现在不同年份间稳定遗传的位点。如, 3年间NPC均可检测到2个显著的SNP位点(SNP_2和SNP_3), 而LPC也可检测到一个显著SNP (SNP_40)。这些结果表明, 虽然香气品质相关性状的遗传基础较为复杂, 但利用多年份鉴定的方法仍可以有效挖掘到稳定遗传的基因位点。
前人研究表明, 香气品质通常由多基因控制, 是受遗传因素及环境互作等多方面影响的数量性状[16]。全基因组关联分析可以有效地鉴定出调控性状的关键位点和候选基因, 具有较高的分辨率, 为研究复杂农艺性状的遗传机制提供了更多线索[17-18]。基于GWAS, 从305份草莓群体中确定了62个与挥发性香气物质有关的信号簇, 并对其物理位置和亚基因组信息进行了基本阐述, 通过开发相对应的分子标记, 可进行辅助选择, 达到改善草莓风味的效果[19]。对321份番茄群体中的结构变异(structural variations, SVs)进行基因分型, 基于GWAS在10号染色体上发现一个347 bp的缺失变异, 该变异与番茄果实中香叶酮的含量密切相关[20]。Wang等[21]通过多组学相结合的方式, 在萜类合成酶的基因中发现了结构变异, 并对乌龙茶高香气特征的遗传基础进行了剖析。本研究利用169个茶树自然杂交后代单株进行NPC与LPC的全基因组关联分析, 共检测到显著关联位点50个, 其中共定位SNP位点1个, 说明NPC与LPC间的调控机制可能存在较大的差异。
3.2 香气品质相关性状候选基因预测
基于混合线性模型、位点注释信息及相关文献报道, 本研究挖掘出位于SNP位点连锁区间内最可能的候选基因, 共59个。这些基因包括糖基转移酶蛋白、糖苷水解酶蛋白、CYP450蛋白、BAHD酰基转移酶蛋白、MYB转录因子、NAC转录因子、WRKY转录因子以及参与萜类化合物生物合成途径的关键酶基因。植物的香味通常与游离的挥发性物质有关, 在糖基转移酶(glycosyltransferases, GTs)的作用下以糖苷形式在植物中贮存[22]。在葡萄中, 糖基转移酶基因VvGT7和VvGT14编码的蛋白酶以香叶醇、芳樟醇等萜类香气成分做底物, 2个基因的表达水平与果实中糖苷的积累量相关[23]。糖苷水解酶(glycoside hydrolases, GHs)是茶叶的一种内源酶, 通常位于细胞质中, 能水解糖苷类香气前体物质, 是释放茶叶香气物质的有效工具[24]。由此推测, 糖基转移酶和糖苷水解酶蛋白可通过控制萜烯类糖苷的合成与水解, 从而调控游离态橙花叔醇和芳樟醇的贮存与释放, 最终影响茶叶香气品质。
与可可和咖啡相比, 茶树中存在着发生了快速扩张的基因家族, 其中TPS被认为是与茶叶香气有关的一种重要酶, 其家族成员的扩张有利于提高茶叶的香气品质[25]。有研究发现, TPS基因在葡萄的果实中表达, 与葡萄成熟过程中香气物质的合成积累有关[26]。拟南芥萜类合酶与细胞色素P450编码的基因与在花器官形成以及开花期紧密共表达,可激活, 而则可产生多种萜烯类挥发性香气物质[27]。在鸡蛋花中, 基因编码的细胞色素P450家族蛋白, 可催化多种VOCs的生物合成, 在烟草花中过表达亦可导致VOCs的大量释放[28]。由此推测, CYP450可通过激活TPS基因的表达, 进而催化橙花叔醇与芳樟醇的生物合成与大量释放。
目前, 越来越多的研究表明, WRKY、MYB、NAC等转录因子参与植物次级代谢产物萜类物质的合成代谢调控, 在植物挥发性香气物质的研究中发挥着重要的作用。如番茄WRKY转录因子过表达, 可显著提高萜烯合成MEP途径中相关基因的表达水平, 而CRISPR/Cas9-slwrky35基因敲除的番茄果实中, MEP途径相关基因的表达水平及其萜类物质的合成显著降低[29]。果实成熟过程中, 芳香物质的合成会受转录因子NAC的调控, 且该分子机制在番茄、桃子、苹果以及猕猴桃等多种植物中保守存在[30-32]。而MYB转录因子则与蝴蝶兰[33]、蕙兰[34]以及草莓果实[35]的香气形成有关, 可通过激活下游合成途径中关键结构基因的启动子表达, 促进香气物质的生物合成。由此推测, 茶树WRKY、MYB、NAC等转录因子有可能通过激活VOCs合成基因启动子元件的方式调控其合成基因的转录和表达。
此外, 本研究还发现了一个一因多效位点SNP_ 39, 该位点优异等位变异可显著提高NPC与LPC, 故挖掘该位点连锁区域的候选基因对茶树香气类型的改良具有重要意义。该位点候选基因编码植物特有的BAHD酰基转移酶, 是参与多种次生代谢物如萜类、花青素等合成修饰的重要酶类之一[36]。蔷薇科水果梨的BAHD酰基转移酶家族成员与其果实的香气生物合成密切相关[11]; 拟南芥BAHD酰基转移酶基因与参与萜类化合物的合成修饰[37]。由此推测,可能通过调控萜类化合物的合成修饰从而影响茶树的香气品质, 其具体的调控机制尚有待进一步的证实。综上所述, 本研究挖掘到的与香气性状相关的候选基因, 为后续基因的精细定位、基因克隆以及香气性状遗传机理的解析提供了参考。
3.3 分子标记辅助选择在未来育种中的应用
分子标记辅助选择能够加速育种进程, 提高选择效率, 是植物育种技术的有力工具[38]。Eggink等[39]基于感官评价与代谢分析, 在辣椒的种间回交群体中定位了果实的萜类化合物QTL位点, 对培育独特风味的辣椒新品种起理论指导意义。通过全基因组关联分析可以挖掘到与目标性状显著关联的标记位点, 有助于分子标记的开发与利用。本研究所选群体的母本材料FT516与FT104是高香型的优异种质, 研究结果可应用于FT516与FT104自然杂交后代的群体改良与品种选育。对主效位点进行有利等位变异分析, 发现与NPC和LPC同时关联的位点SNP_39 (Sca.1970: 1955068)的有利等位基因型均为AA, 且不同基因型间的表型差异达到极显著水平, 基于该变异开发与NPC和LPC相关的分子标记, 或可达到多性状同时改良的效果。
此外, 针对该类有利等位变异开发分子标记, 还可在实际的选育工作中进行有利等位基因的选择或累加[40]。本研究对主效位点深入分析发现, 4个优异等位变异位点对茶树NPC和LPC皆具有显著调控效应, 有较大的应用前景, 可作为茶树香气品质遗传改良的重要位点。例如SNP_3在不同年份间均与NPC关联且表型贡献率较高, 其有利等位变异的基因型为CC, 在育种实践中可定向选择该基因型达到提高NPC的效果。
4 结论
本研究以169个茶树自然杂交后代单株作为关联群体, 利用个675,245个SNP标记, 采用MLM模型针对NPC和LPC进行全基因组关联分析, 共检测到50个显著关联的SNP位点, 其中在2个及其以上环境中被稳定检测到的有28个。针对2个目标性状, 共鉴定出4个主效SNP位点, 其中1个为可同时调控NPC和LPC的一因多效位点。在显著SNP位点上下游各100 kb的区间内, 筛选出最可能的候选基因共59个, 主要涉及茶树的糖类代谢、转录调控、萜烯类生物合成等多个生物学过程, 其中26个基因在绿茶品种‘福云6号’与乌龙茶品种‘悦茗香’中的表达水平存在显著差异。本研究结果可为进一步揭示茶树NPC和LPC的遗传机制, 以及高香型茶树新品种选育提供参考。
[1] Wang D, Yoshimura T, Kubota K, Kobayashi A. Analysis of glycosidically bound aroma precursors in tea leaves: I. Qualitative and quantitative analyses of glycosides with aglycons as aroma compounds., 2000, 48: 5411–5418.
[2] Liu G F, Liu J J, He Z R, Wang F M, Yang H, Yan Y F, Gao M J, Gruber M, Wan X C, Wei S. Implementation of CsLIS/NES in linalool biosynthesis involves transcript splicing regulation in.,2018, 41: 176–186.
[3] Wei K, Wang X, Hao X, Qian Y, Li X, Xu L, Ruan L, Wang Y, Zhang Y, Bai P, Li Q, Aktar S, Hu X, Zheng G, Wang L, Liu B, He W, Cheng H, Wang L. Development of a genome-wide 200K SNP array and its application for high-density genetic mapping and origin analysis of.,2022, 20: 414–416.
[4] Cao K, Yang X, Li Y, Zhu G, Fang W, Chen C, Wang X, Wu J, Wang L. New high-quality peach (L. Batsch) genome assembly to analyze the molecular evolutionary mechanism of volatile compounds in peach fruits.,2021, 108: 281–295.
[5] Gong C, He N, Zhu H, Anees M, Lu X, Liu W. Multi-omics integration to explore the molecular insight into the volatile organic compounds in watermelon.,2023, 166: 112603.
[6] Ferrão L F V, Johnson T S, Benevenuto J, Edger P P, Colquhoun T A, Munoz P R. Genome-wide association of volatiles reveals candidate loci for blueberry flavor.,2020, 226: 1725–1737.
[7] 严威凯. 品种选育与评价的原理和方法评述. 作物学报, 2022, 48: 2137–2154. Yan W K. A critical review on the principles and procedures for cultivar development and evaluation., 2022, 48: 2137–2154 (in Chinese with English abstract).
[8] Xia E H, Li F D, Tong W, Li P H, Wu Q, Zhao H J, Ge R H, Li R P, Li Y Y, Zhang Z Z, Wei C L, Wan X C. Tea Plant Information Archive (TPIA): a comprehensive genomics and bioinformatics platform for tea plant.,2019, 17: 1938–1953.
[9] 王让剑, 杨军, 张力岚, 高香凤. 茶树新梢中香叶醇樱草糖苷含量的全基因组关联分析. 作物学报,2023, 49: 1843–1859. Wang R J, Yang J, Zhang L L, Gao X F. Genome-wide association analysis of geraniol primrose glycoside abundance in tender tea shoots., 2023, 49: 1843–1859 (in Chinese with English abstract).
[10] Chen J D, He W Z, Chen S, Chen Q Y, Ma J Q, Jin J Q, Ma C L, Moon D G, Ercisli S, Yao M Z, Chen L. TeaGVD: a comprehensive database of genomic variations for uncovering the genetic architecture of metabolic traits in tea plants., 2022, 13: 1056891.
[11] Liu C, Qiao X, Li Q, Zeng W, Wei S, Wang X, Chen Y, Wu X, Wu J, Yin H, Zhang S. Genome-wide comparative analysis of thesuperfamily in seven Rosaceae species and expression analysis in pear ().,2020, 20: 14.
[12] Fang K, Xia Z, Li H, Jiang X, Qin D, Wang Q, Wang Q, Pan C, Li B, Wu H. Genome-wide association analysis identified molecular markers associated with important tea flavor-related metabolites.,2021, 8: 42.
[13] Hazra A, Kumar R, Sengupta C, Das S. Genome-wide SNP discovery from Darjeeling tea cultivars: their functional impacts and application toward population structure and trait associations.,2021, 113: 66–78.
[14] Huang R, Wang J Y, Yao M Z, Ma C L, Chen L. Quantitative trait loci mapping for free amino acid content using an albino population and SNP markers provides insight into the genetic improvement of tea plants.,2022, 9: uhab029.
[15] Yamashita H, Uchida T, Tanaka Y, Katai H, Nagano A J, Morita A, Ikka T. Genomic predictions and genome-wide association studies based on RAD-seq of quality-related metabolites for the genomics-assisted breeding of tea plants.,2020, 10: 17480.
[16] Heng Z, Xu X, Xu X, Wang H, Liu L, Li Z, Li Z, You Q, Sun B, Gong C, Yin Y, Li Y, Li T. Characterization of odor-contributing volatile in‘JT-1’ fruits during development and transcriptome analysis of key fruit-aroma formation periods.,2023, 309: 111691.
[17] 马雅杰, 鲍建喜, 高悦欣, 李雅楠, 秦文萱, 王彦博, 龙艳, 李金萍, 董振营, 万向元. 玉米株高和穗位高性状全基因组关联分析. 作物学报, 2023, 49: 647–661. Ma Y J, Bao J X, Gao Y X, Li Y N, Qin W X, Wang Y B, Long Y, Li J P, Dong Z Y, Wan X Y. Genome-wide association analysis of plant height and ear height related traits in maize., 2023, 49: 647–661 (in Chinese with English abstract).
[18] 董一帆, 任毅, 程宇坤, 王睿, 张志辉, 时晓磊, 耿洪伟. 冬小麦籽粒主要品质性状的全基因组关联分析. 中国农业科学,2023, 56: 2047–2063. Dong Y F, Ren Y, Cheng Y K, Wang R, Zhang Z H, Shi X L, Geng H W. Genome-wide association study of grain main quality related traits in winter wheat., 2023, 56: 2047–2063 (in Chinese with English abstract).
[19] Fan Z, Tieman D M, Knapp S J, Zerbe P, Famula R, Barbey C R, Folta K M, Amadeu R R, Lee M, Oh Y, Lee S, Whitaker V M. A multi-omics framework reveals strawberry flavor genes and their regulatory elements.,2022, 236: 1089–1107.
[20] Li N, He Q, Wang J, Wang B, Zhao J, Huang S, Yang T, Tang Y, Yang S, Aisimutuola P, Xu R, Hu J, Jia C, Ma K, Li Z, Jiang F, Gao J, Lan H, Zhou Y, Zhang X, Huang S, Fei Z, Wang H, Li H, Yu Q. Super-pangenome analyses highlight genomic diversity and structural variation across wild and cultivated tomato species.,2023, 55: 852–860.
[21] Wang P, Yu J, Jin S, Chen S, Yue C, Wang W, Gao S, Cao H, Zheng Y, Gu M, Chen X, Sun Y, Guo Y, Yang J, Zhang X, Ye N. Genetic basis of high aroma and stress tolerance in the oolong tea cultivar genome.,2021, 8: 107.
[22] Bönisch F, Frotscher J, Stanitzek S, Ruehl E, Wüst M, Bitz O, Schwab W. A UDP-glucose: monoterpenol glucosyltransferase adds to the chemical diversity of the grapevine metabolome.,2014, 165: 561–581.
[23] Li X Y, Wen Y Q, Meng N, Qian X, Pan Q H. Monoterpenyl glycosyltransferases differentially contribute to production of monoterpenyl glycosides in two aromaticvarieties.,2017, 8: 1226.
[24] Rodriguez-Bencomo J J, Muñoz-González C, Andujar-Ortiz I, Martín-Álvarez P J, Moreno-Arribas M V, Pozo-Bayón M Á. Assessment of the effect of the non-volatile wine matrix on the volatility of typical wine aroma compounds by headspace solid phase microextraction/gas chromatography analysis.,2011, 91: 2484–2494.
[25] Xia E H, Tong W, Hou Y, An Y, Chen L, Wu Q, Liu Y L, Yu J, Li F, Li R, Li P, Zhao H, Ge R, Huang J, Mallano H I, Zhang Y, Liu S, Deng W, Song C, Zhang Z, Zhao J, Wei S, Zhang Z, Xia T, Wei C, Wan X. The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into genome evolution and adaptation of tea plants.,2020, 13: 1013–1026.
[26] Martin D, Aubourg S, Schouwey M, Daviet L, Schalk M, Toub O, Lund S, Bohlmann J. Functional annotation, genome organization and phylogeny of the grapevine () terpene synthase gene family based on genome assembly, FLcDNA cloning, and enzyme assays.,2010, 10: 226.
[27] Boachon B, Burdloff Y, Ruan J X, Rojo R, Junker R R, Vincent B, Nicolè F, Bringel F, Lesot A, Henry L, Bassard J E, Mathieu S, Allouche L, Kaplan I, Dudareva N, Vuilleumier S, Miesch L, André F, Navrot N, Chen X Y, Werck-Reichhart D. A promiscuous CYP706A3 reduces terpene volatile emission from Arabidopsis flowers, affecting florivores and the floral microbiome.,2019, 31: 2947–2972.
[28] Dhandapani S, Jin J, Sridhar V, Chua N H, Jang I C. CYP79D73 participates in biosynthesis of floral scent compound 2-phenylethanol in.,2019, 180: 171–184.
[29] Yuan Y, Ren S, Liu X, Su L, Wu Y, Zhang W, Li Y, Jiang Y, Wang H, Fu R,Bouzayen M, Liu M, Zhang Y. SlWRKY35 positively regulates carotenoid biosynthesis by activating the MEP pathway in tomato fruit.,2022, 234: 164–178.
[30] Cao X, Wei C, Duan W, Gao Y, Kuang J, Liu M, Chen K, Klee H, Zhang B. Transcriptional and epigenetic analysis reveals that NAC transcription factors regulate fruit flavor ester biosynthesis.,2021, 106: 785–800.
[31] Gao Y, Lin Y, Xu M, Bian H, Zhang C, Wang J, Wang H, Xu Y, Niu Q, Zuo J, Fu D Q, Pan Y, Chen K, Klee H, Lang Z, Zhang B. The role and interaction between transcription factor NAC-NOR and DNA demethylase SlDML2 in the biosynthesis of tomato fruit flavor volatiles.,2022, 235: 1913–1926.
[32] Wang R, Shu P, Zhang C, Zhang J, Chen Y, Zhang Y, Du K, Xie Y, Li M, Ma T, Zhang Y, Li Z, Grierson D, Pirrello J, Chen K, Bouzayen M, Zhang B, Liu M. Integrative analyses of metabolome and genome-wide transcriptome reveal the regulatory network governing flavor formation in kiwifruit ().,2022, 233: 373–389.
[33] Hsiao Y Y, Tsai W C, Kuoh C S, Huang T H, Wang H C, Wu T S, Leu Y L, Chen W H, Chen H H. Comparison of transcripts inand(Orchidaceae) flowers to deduce monoterpene biosynthesis pathway.,2006, 6: 14.
[34] Xu Y, Zhou J, Lu S, Wang S, Zhou Y. Cloning and molecular characterization ofassociated with the regulation of methyl jasmonate biosynthesis in.,2020, 89: 593–601.
[35] Wang S, Shi M, Zhang Y, Pan Z, Xie X, Zhang L, Sun P, Feng H, Xue H, Fang C, Zhao J. The R2R3-MYB transcription factor FaMYB63 participates in regulation of eugenol production in strawberry.,2022, 188: 2146–2165.
[36] Srivastava S, Sangwan R S. Analysis oftranscriptome for BAHD alcohol acyltransferase genes: identification and diversity of expression in leaf, stem and root.,2012, 21: 108–118.
[37] Wang M, Liu X, Wang R, Li W, Rodermel S, Yu F. Overexpression of a putativeBAHD acyltransferase causes dwarfism that can be rescued by brassinosteroid.,2012, 63: 5787–5801.
[38] Bueren E T, Østergård H, Vriend H, Backes G. The role of molecular markers and marker assisted selection in breeding for organic and low-input agriculture.,2010, 175: 51–64.
[39] Eggink P M, Tikunov Y, Maliepaard C, Haanstra J P, de Rooij H, Vogelaar A, Gutteling E W, Freymark G, Bovy A G, Visser R G. Capturing flavors fromby introgression in sweet pepper., 2014, 127: 373–390.
[40] 王慧玲, 闫爱玲, 王晓玥, 刘振华, 任建成, 徐海英, 孙磊. 葡萄果粒质量相关性状全基因组关联分析. 中国农业科学, 2023, 56: 1561–1573.Wang H L, Yan A L, Wang X Y, Liu Z H, Ren J C, Xu H Y, Sun L. Genome-wide association studies for grape berry weight related traits., 2023, 56: 1561–1573(in Chinese with English abstract).
附表1 169份茶树自然杂交后代单株名称及其信息来源
Table S1 Names and origin information of 169 natural hybrid progenies
序号No.名称Name母本来源Maternal parent序号No.名称Name母本来源Maternal parent 104-1FT104(♀)8616-15FT516(♀) 204-10FT104(♀)8716-16FT516(♀) 304-11FT104(♀)8816-17FT516(♀) 404-12FT104(♀)8916-18FT516(♀) 504-13FT104(♀)9016-19FT516(♀) 604-14FT104(♀)9116-2FT516(♀) 704-15FT104(♀)9216-20FT516(♀) 804-16FT104(♀)9316-21FT516(♀) 904-17FT104(♀)9416-22FT516(♀) 1004-18FT104(♀)9516-23FT516(♀) 1104-19FT104(♀)9616-24FT516(♀) 1204-2FT104(♀)9716-25FT516(♀) 1304-20FT104(♀)9816-26FT516(♀) 1404-21FT104(♀)9916-27FT516(♀) 1504-22FT104(♀)10016-28FT516(♀) 1604-23FT104(♀)10116-29FT516(♀) 1704-24FT104(♀)10216-3FT516(♀) 1804-25FT104(♀)10316-30FT516(♀) 1904-26FT104(♀)10416-31FT516(♀) 2004-27FT104(♀)10516-32FT516(♀) 2104-28FT104(♀)10616-33FT516(♀) 2204-29FT104(♀)10716-34FT516(♀) 2304-3FT104(♀)10816-35FT516(♀) 2404-30FT104(♀)10916-36FT516(♀) 2504-31FT104(♀)11016-37FT516(♀) 2604-32FT104(♀)11116-38FT516(♀) 2704-33FT104(♀)11216-39FT516(♀) 2804-34FT104(♀)11316-4FT516(♀) 2904-35FT104(♀)11416-40FT516(♀) 3004-36FT104(♀)11516-41FT516(♀) 3104-37FT104(♀)11616-42FT516(♀) 3204-38FT104(♀)11716-43FT516(♀) 3304-39FT104(♀)11816-44FT516(♀) 3404-4FT104(♀)11916-45FT516(♀) 3504-40FT104(♀)12016-46FT516(♀) 3604-41FT104(♀)12116-47FT516(♀) 3704-42FT104(♀)12216-48FT516(♀) 3804-43FT104(♀)12316-49FT516(♀) 3904-44FT104(♀)12416-5FT516(♀) 4004-45FT104(♀)12516-50FT516(♀) 4104-46FT104(♀)12616-51FT516(♀) 4204-47FT104(♀)12716-52FT516(♀) 4304-48FT104(♀)12816-53FT516(♀) 4404-49FT104(♀)12916-54FT516(♀)
(续附表1)
序号No.名称Name母本来源Maternal parent序号No.名称Name母本来源Maternal parent 4504-5FT104(♀)13016-55FT516(♀) 4604-50FT104(♀)13116-56FT516(♀) 4704-51FT104(♀)13216-57FT516(♀) 4804-52FT104(♀)13316-58FT516(♀) 4904-53FT104(♀)13416-59FT516(♀) 5004-54FT104(♀)13516-6FT516(♀) 5104-55FT104(♀)13616-60FT516(♀) 5204-56FT104(♀)13716-61FT516(♀) 5304-57FT104(♀)13816-62FT516(♀) 5404-58FT104(♀)13916-63FT516(♀) 5504-59FT104(♀)14016-64FT516(♀) 5604-6FT104(♀)14116-65FT516(♀) 5704-60FT104(♀)14216-66FT516(♀) 5804-61FT104(♀)14316-67FT516(♀) 5904-62FT104(♀)14416-68FT516(♀) 6004-63FT104(♀)14516-69FT516(♀) 6104-64FT104(♀)14616-7FT516(♀) 6204-65FT104(♀)14716-70FT516(♀) 6304-66FT104(♀)14816-71FT516(♀) 6404-67FT104(♀)14916-72FT516(♀) 6504-68FT104(♀)15016-73FT516(♀) 6604-69FT104(♀)15116-74FT516(♀) 6704-7FT104(♀)15216-75FT516(♀) 6804-70FT104(♀)15316-76FT516(♀) 6904-71FT104(♀)15416-77FT516(♀) 7004-72FT104(♀)15516-78FT516(♀) 7104-73FT104(♀)15616-79FT516(♀) 7204-74FT104(♀)15716-8FT516(♀) 7304-75FT104(♀)15816-80FT516(♀) 7404-76FT104(♀)15916-81FT516(♀) 7504-77FT104(♀)16016-82FT516(♀) 7604-78FT104(♀)16116-83FT516(♀) 7704-79FT104(♀)16216-84FT516(♀) 7804-8FT104(♀)16316-85FT516(♀) 7904-9FT104(♀)16416-86FT516(♀) 8016-1FT516(♀)16516-87FT516(♀) 8116-10FT516(♀)16616-88FT516(♀) 8216-11FT516(♀)16716-89FT516(♀) 8316-12FT516(♀)16816-9FT516(♀) 8416-13FT516(♀)16916-90FT516(♀) 8516-14FT516(♀)
FT516、FT104是“十二五”国家科技支撑计划项目课题任务“福建茶树种质鉴定评价与新品种选育(2011BAD01B01-FJCKS)”鉴定的10个优异种质中制茶品质得分居前2位的高香型优异种质。
FT516 and FT104 are the top 2 high aroma excellent tea varieties among the 10 excellent germplasm identified in the “Fujian Tea Germplasm Identification and Evaluation and New Variety Selection (2011BAD01B01-FJCKS)” project of the National Science and Technology Support Plan for the 12th Five-Year Plan.

附图1 GWAS候选基因在茶树各组织中的表达
Fig. S1 Expression profiles of candidate genes in different tissues of tea by GWAS
Genome-wide association study and candidate gene prediction of nerolidol and linalool primeveroside content in tea plants
ZHANG Li-Lan1,2, YANG Jun1,2, and WANG Rang-Jian1,2,*
1Tea Research Institute, Fujian Academy of Agricultural Sciences, Fuzhou 350013, Fujian, China;2Fujian Branch, National Center for Tea Improvement, Fuzhou 350013, Fujian, China
Nerolidol and linalool are volatile terpene alcohols compounds widely distributed in plants. They are mainly existing in the form of primeveroside in tea plant tender shoots, and increasing their content is of great significance for improving the aroma quality of tea. The objective of this study is to reveal the genetic mechanism of nerolidol and linalool primeveroside in tea plants. 169 natural hybrid progenies were used as associated populations, and the contents of nerolidol and linalool primeveroside in tea plant tender shoots in three years were analyzed by using 675,245 single nucleotide polymorphism (SNP) markers evenly distributed uniformly on the chromosomes of tea genome. The results showed that the phenotypic variation of nerolidol and linalool primeveroside content were 60.83%–80.08%, and the broad-sense heritability were 51.29% and 61.87% respectively. Nerolidol and linalool primeveroside content were in normal distribution, suggesting that the traits have typical genetic characteristics of quantitative traits. A total of 50 significantly associated loci were detected by GWAS, and each locus contributed more than 20% to the variations of nerolidol and linalool primeveroside content, of which the maximum contribution rate of nerolidol primeveroside content (NPC) variation site was 38.73%, and the maximum contribution rate of linalool primeveroside content (LPC) variation site was 39.07%. Furthermore, the elite alles of the four major SNPs was identified by allelic variation effect analysis, among which one locus that could affected NPC and LPC simultaneously. Finally, a total of 59 genes were annotated in the confidence intervals of each significantly associated loci, and the most likely candidate genes were predicted according to the comparison with previous reports and gene functional annotations. These candidate genes were mainly involved in multiple biological processes such as sugar metabolism, transcriptional regulation, terpene biosynthesis. Among them, there were significant differences in the relative expression levels of 26 genes between green tea varieties and oolong tea varieties. This study provides new information for further dissecting the genetic mechanism of nerolidol and linalool primeveroside content in tea plants, and provides important gene resources for accelerating the breeding of new tea varieties with high quality.
; nerolidol primeveroside; linalool primeveroside; genome-wide association study; candidate gene
10.3724/SP.J.1006.2024.34124
本研究由福建省科技计划项目(2023R1090)和福建省农业科学院科技专项(ZYTS202408)资助。
This study was supported by the Technology Plan Project of Fujian Province (2023R1090) and the Science and Technology Specific Project of Fujian Academy of Agricultural Science (ZYTS202408).
王让剑, E-mail: wangrj@faas.cn
E-mail: lilanzhang0114@foxmail.com
2023-07-18;
2023-10-23;
20213-11-13.
URL: https://link.cnki.net/urlid/11.1809.S.20231110.0856.004
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
猜你喜欢
小读者之友(2024年1期)2024-03-29
青岛科技大学学报(自然科学版)(2022年3期)2022-07-02
广州化工(2021年19期)2021-10-25
茶业通报(2020年1期)2020-12-18
小学生学习指导·爆笑校园(2019年2期)2019-09-10
中国调味品(2017年2期)2017-03-20
广西林业科学(2016年2期)2016-03-20
食品界(2016年4期)2016-02-27
物理化学学报(2015年7期)2015-12-30
经济林研究(2015年3期)2015-12-21
