
大豆类病变皱叶突变体NT301遗传分析和2对基因定位
2024-03-28 02:36:30王亚琪徐海风李曙光傅蒙蒙余希文赵志鑫杨加银赵团结
作物学报 2024年4期
王亚琪 徐海风 李曙光 傅蒙蒙 余希文 赵志鑫 杨加银,* 赵团结
大豆类病变皱叶突变体遗传分析和2对基因定位
王亚琪1徐海风1李曙光1傅蒙蒙1余希文1赵志鑫1杨加银1,*赵团结2,*
1江苏徐淮地区淮阴农业科学研究所 / 淮安市农业生物技术重点实验室 / 农业农村部淮河下游种质创制重点实验室, 江苏淮安 223001;2南京农业大学大豆研究所/ 国家大豆改良中心(南京) / 农业农村部大豆生物学与遗传育种重点实验室(综合) / 作物遗传与种质创新国家重点实验室, 江苏南京 210095
通过研究类病变突变体, 挖掘抗病基因, 利用分子设计育种的方法快速培育优良抗病大豆新品种, 可减轻化学农药对环境的污染, 降低病害的抗药性。本研究以60Coγ诱变获得的类病变皱叶突变体为父本, 分别与W82、KF1和KF35进行杂交, 构建了F2和F2:3分离群体, 通过SSR标记和SNP分析, 将目标基因1 ()缩小到18号染色体937 kb区间内, 包含66个候选基因, 将目标基因2 ()缩小到8号染色体130 kb区间内, 包含15个候选基因。接着, 本研究利用基因芯片技术对近等基因系进行了基因表达谱研究, 得到了差异表达基因参与的KEGG调控通路。另外对8号染色体的15个候选基因进行半定量与荧光定量RT-PCR表达分析发现, 只有基因在突变体与野生型中的差异达到了4倍, 推测基因是突变体的候选基因。
大豆; 类病变突变体; 皱叶; 基因表达谱
大豆含有丰富优质的蛋白和油脂, 是重要的粮食作物和经济作物, 在我国粮食和饲料结构中占有重要位置。而我国大豆生产正面临严峻的形势, 随着对大豆需求逐年增加, 供求矛盾日益突出, 如今已经成为世界最大的大豆进口国[1-2]。病害严重影响大豆产量和品质, 培育优良抗病品种, 既节约了资源, 又可减少化学农药对环境的污染, 还能降低病害的抗药性。而分子设计育种是解决这一困境的重要途径, 加强基础研究, 挖掘重要性状的关键基因及其调控网络, 是分子设计育种的关键。
近年来的许多研究表明, 类病变突变体病斑的形成与过敏反应及程序性细胞死亡有关, 是研究植物抗病分子机制的良好材料。类病变突变体是指植物组织上自发形成细胞死亡, 与外部环境的胁迫没有直接关系, 往往形成病斑或过敏反应[3-4]。1997年, 研究人员首先在玉米和拟南芥中鉴定到了控制类病变性状的基因[5-6], 后来在番茄和水稻中也鉴定到了类病变的候选基因[7-8]。Wang等[9]通过图位克隆的方法从大豆类病变突变体中鉴定出候选基因脂氢过氧化物裂解酶HPL, 能够响应病虫害胁迫。Ma等[10]鉴定出一个大豆光依赖的类病变突变体, 其候选基因编码一种共型卟啉原III氧化酶GmLMM2, 参与四吡咯生物合成, 突变体增强了对大豆疫霉的抗性。这些研究揭示了大多数类病变突变体能够激活防御反应、甚至增强了对病原菌的抗性, 这些基因涉及多个途径: 如抗病信号途径、植物激素及防卫信号分子、程序性细胞死亡失控、叶绿素合成和脂类代谢等。
类病变皱叶突变体主要特点为叶脉与叶肉细胞发育不协调, 产生与病毒感染相似的形态特征, 叶面积大大减小, 结实率降低, 而大豆中的研究并不深入。Stephens等[11]从再生植株中发现了一个不稳定遗传的皱叶突变体, 后代分离不稳定, 属于细胞质遗传, 往往出现嵌合体。Wilcox和Abney[12]从M2中鉴定到一个隐性遗传的叶宽变窄的皱叶突变体, 遗传解析表明由单基因控制。聂智星等[13]从60Coγ诱变的大豆突变体库中筛选到一个皱叶突变体, 叶缘卷曲, 类似花叶病毒感染。在普通菜豆(L.)中, 异源杂交后代出现了双隐性基因控制的皱叶突变体[14]。Song等[15]鉴定到一个大豆皱叶突变体, 小叶顶端坏死, 而小叶其他部分继续生长, 叶面积和叶片干重显著降低。Ochar等[16]报道了一个EMS诱变的大豆皱叶突变体, 编码具有CCT结构域的B-box型锌指蛋白突变导致叶片皱缩, 株高降低。
基因重复或多倍化在植物进化过程中普遍存在,且不同的进化阶段都有发生。全基因组复制是重复基因最重要的来源之一, 其中一部分基因消亡, 一部分基因被保留但功能发生了变化, 这些对进化起了重要作用[17]。大豆基因组在过去的6000万年时间里至少经历了2轮全基因组复制, 存在大量的重复序列, 研究者推测这些重复基因的功能也发生了变化[18], 研究重复基因的功能对研究进化起到了重要作用。之前的研究中, 我们发现了一个新的类病变突变体, 叶片皱缩, 株高降低, 对F2代和F2:3家系的遗传分析表明该性状被2对隐性基因控制, 功能冗余, 并且将这2对基因定位在18号和8号染色体上。
本研究将目标基因1 ()缩小到18号染色体937 kb区间, 目标基因2 ()缩小到8号染色体130 kb区间, 包含15个候选基因。为进一步缩小候选基因的范围, 本研究对8号染色体的15个候选基因进行半定量与实时荧光定量表达分析发现, 只有基因在突变体与野生型中的差异达到了4倍, 推测基因是突变体的候选基因。
1 材料与方法
1.1 遗传材料获得与构建
大豆品种玉誉(Tamahomore)、Williams 82 (W82)、科丰1号(Kefeng 1, KF1)、科丰35 (Kefeng 35, KF35)等种植资源均来自于南京农业大学国家大豆改良中心。突变体来自于60Coγ诱变的玉誉[19],并经过多代回交及自交, 得到近等基因系的野生型(wild type, WT)和突变型(mutant type, MT)。然后分别以W82、KF1和KF35为母本, 以为父本构建杂交群体, W82×杂交组合F2代分离群体共1912株, F2:3代株系共156行; KF1×杂交组合F2代分离群体共451株, F2:3代株系共182行; KF35×杂交组合F2代分离群体共362株, F2:3代株系共176行。
1.2 石蜡切片观察叶脉结构
在V4至V5时期, 采集野生型和突变体植株顶部比较幼嫩的叶片, 采用常规石蜡切片法制片。用手术刀片切成大小0.5~1.0 cm左右的组织, FAA固定液固定, 保证组织表面无气泡, 1 d后转移至70%乙醇中, 按照不同浓度的乙醇脱水, 然后用二甲苯透明剂透明, 用石蜡浸蜡和包埋, 切片机切片, 粘附剂粘片与烤片, 再用二甲苯脱蜡, 不同浓度梯度的酒精复水, 用番红-固绿对染, 再经过不同浓度梯度的酒精脱水、二甲苯透明和中性树胶封固, 在LEICA DMLB型显微镜下观察并照相。
1.3 目的基因定位
对于质量性状, 根据孟德尔遗传定律来进行遗传分析。由于突变体第1片三出复叶就表现皱叶, 肉眼很容易分辨, 因此, 用肉眼观测进行表型调查。调查F1、F2和F2:3代(F2单株衍生的株行)的表型, 统计其性状分离比, 用卡平方适合性测验计算其显著性。以隐性遗传为例, 如果F1代单株全部表现为正常, F2代单株出现正常︰皱叶=3︰1, 表现正常F2单株衍生的F2:3代株行正常不分离行︰分离行=1︰2, 则为隐性单基因遗传。如果F1代单株全部表现为正常, F2代单株出现正常︰皱叶=15︰1, 表现正常F2单株衍生的F2:3代株行正常不分离行︰分离行=7︰8, 则为隐性双基因遗传。
采用SSR标记和SNP连锁定位的方法进行基因定位。对F2和F2:3群体, V3至V4期突变体皱叶表型出现时, 调查表型并取顶部幼嫩叶片, 用CTAB法提取DNA。根据Song等[20]开发的大豆全基因组微卫星标记(SSR)对初定位区间进行标记加密, 当定位区间还有交换单株时, 就要开发新的SSR标记, 进一步缩小定位区间。方法如下: 在SoyBase (https:// soybase.org/)中下载W82参考基因组目标区段序列, 利用SSRHunter[21]筛选SSR标记, 参数设置优先选择3~6个碱基的重复序列, 并根据软件所给的结果, 利用NCBI Primer-BLAST (https://www.ncbi.nlm.nih. gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome)在上下游150 bp内设计引物, 保证扩增产物长度在100~300 bp之间, 上下游引物m值相差不大于5℃, 由通用生物(安徽)股份有限公司合成。PCR扩增利用2×Master Mix (P112-01, 诺唯赞生物科技股份有限公司, 南京)进行, 用8%的聚丙烯酰胺凝胶(PAGE)对PCR产物进行电泳检测和银染显色[19]。
1.4 基因表达谱试验流程及数据分析
当突变体出现皱叶表型时, 混样取野生型和突变体的叶片, 迅速装入冻存管, 并置于液氮中, 以备提取大豆总RNA, 野生型和突变体各3个生物学重复, 送上海伯豪生物公司进行基因表达谱分析。流程是: 首先, 将检测合格的RNA样本进行纯化, 再反转录合成第1链、第2链cDNA, 然后利用双链cDNA合成生物素标记的aRNA, 纯化后与芯片杂交,进行测序分析。
1.5 候选基因预测及半定量、定量RT-PCR检测
在SoyBase网站下载定位区间内的所有基因ID, 根据Phytozome (http://phytozome-next.jgi.doe.gov/)和SoyKB (https://soykb.org/)网站的基因功能注释, 预测候选基因, 并下载基因组及转录组的序列, 利用NCBI Primer-BLAST设计半定量和实时荧光定量PCR引物(附表1), 检测定位区间内基因的表达水平。V3至V4时期, 取顶部完全展开的嫩叶(每个材料3个生物学重复), 迅速放入液氮冷冻, 再用总RNA提取试剂盒(DP419, 天根生化科技有限公司, 北京)提取总RNA, 检测其浓度及质量, 并稀释至终浓度为5 ngmL–1, 用反转录试剂盒(R223, 诺唯赞生物科技股份有限公司, 南京)将RNA反转为cDNA。半定量RT-PCR以cDNA为模板, 在普通PCR仪进行, 根据琼脂糖凝胶电泳检测结果确定了内参基因和目的基因以26个循环为最佳。实时荧光定量PCR反应以cDNA为模板, 用染料法试剂盒(Q311, 诺唯赞生物科技股份有限公司, 南京), 在Roche Light Cycler 480 II上进行, 相对表达量采用2–ΔΔCt计算方法。半定量和荧光定量RT-PCR每个样本3个技术重复。
2 结果与分析
2.1 突变体NT301的形态特点与遗传分析
之前的研究已经详细报道了突变体的形态特征[19], 该突变体植株矮小, 结实率低, 叶片表现出感染花叶病毒症状(图1-B, D), 种子大小与野生型无明显差异, 石蜡切片显示皱叶叶片横切面的内部结构(图1-F)。突变体表皮细胞排列紊乱, 不规则凸起, 致使表面形成褶皱, 表皮细胞和维管束之间的厚角组织层数增多, 导致叶脉宽度变窄, 木质部细胞变小, 叶肉细胞单位面积的细胞数目异常增多, 栅栏组织和海绵组织连在一起, 中间几乎没有空隙。田间调查杂交组合后代表型并进行遗传分析。由表1可知, KF1×群体F1代叶片形态表现正常, F2代451个单株中, 有430株表现正常, 21株表现皱叶, 卡方测验符合15︰1的分离比(=0.48), F2:3代182个株系中, 有88个株系表现正常不分离, 94个株系出现分离, 卡方测验符合7︰8的分离比(=0.70)。KF35×群体F1代叶片形态表现正常, F2代362个单株中, 有342株表现正常, 20株表现皱叶, 卡方测验符合15︰1的分离比(=0.80), F2:3代176个株系中, 有87个株系表现正常不分离, 89个株系出现分离, 卡方测验符合7︰8的分离比(=0.51)。已报道过W82×群体F2代单株正常︰皱叶符合15︰1的分离比(=0.57), F2:3代株系正常不分离︰分离符合7︰8的分离比(=0.84)[19]。表明皱叶性状受2对隐性基因控制。

图1 突变体NT301的形态特征
A: 苗期野生型植株; B: 苗期植株; C: 开花期野生型植株; D: 开花期植株; E: 野生型叶片横切面; F:叶片横切面。标尺: 1 cm (A, B); 5 cm (C, D); 200 μm (E, F)。p: 栅栏组织; s: 海绵组织; vb: 维管束。
A: wild-type plants at seedling stage; B: mutantat seedling stage; C: wild-type plants at flowering stage; D: mutantatflowering stage; E: the transverse section of wild-type leaves; F: the transverse section ofleaves. Bar: 1 cm (A, B); 5 cm (C, D); 200 μm (E, F). p: palisade parenchyma; s: spongy parenchyma; vb: vascular bundle.
2.2 突变体NT301皱叶基因定位
在之前的报道中, 将的目标基因定位在了18号染色体BARCSOYSSR_18_0415和BARCSOYSSR_18_0485两个SSR标记之间, 物理距离1.85 Mb, 将目标基因定位在了8号染色体BARCSOYSSR_08_1700和Satt409之间, 物理距离1.18 Mb。为了进一步精细定位, 利用KF1×群体F2:3家系277个隐性单株在18号染色体定位区间内加密了3个标记, 分别是18_0419、18_0444和18_0481, 用这3个标记分别检测出了10个、5个和3个交换单株; 利用W82×群体F2:3家系327个隐性单株加密了2个标记, 分别是18_0419和18_0430, 并检测了标记18_0485, 用这3个标记分别检测出了9个、3个和17个交换单株, 综合2个群体的定位结果, 将定位在了18_0444和18_0481之间, 物理距离937 kb。利用KF1×群体F2:3家系262个隐性单株在8号染色体定位区间内加密了3个标记, 分别是08_1724、08_1736和08_1753, 并检测了标记08_1800, 用这4个标记分别检测出了52个、33个、9个和23个交换单株; 利用W82×群体F2:3家系315个隐性单株加密了5个SSR标记和3个SNP变异位点, 分别是08_1724、08_1738、08_1748、SSR18、SSR40和SNP1、SNP2、SNP3, 用这些标记分别检测出了10个、10个、10个、2个、2个和2个、1个、2个交换单株, 综合2个群体的定位结果, 将定位在了SNP2和SNP3之间, 物理距离130 kb (图2)。根据SoyBase和Phytozome网站的基因注释信息, 18号和8号染色体目标区段内分别有66个和15个候选基因。
2.3 突变体NT301皱叶候选基因参与的调控网络
利用大豆Affymetrix基因组芯片检测皱叶突变体与野生型叶片组织的表达差异, 该芯片包含37,500个大豆()的转录本, 背景值小于100, 检出率为40%以上表明芯片、样本及操作均达到合格标准。在本研究中, 野生型样品1-1、1-2、1-3和突变体样品2-1、2-2、2-3的探针检出率均大于40% (表2), 表明试验数据比较可靠。
采用随机方差模型对差异基因进行筛选, 共获得了3568个差异表达基因, 其中上调2337个, 下调1231个。采用选择Cluster 3.0软件对差异基因进行聚类, 将功能显著差异的600个基因使用hierarchical, average linkage算法进行分析(图3), 野生型和突变体能够明显分为2类, 说明芯片数据的重复性较好。
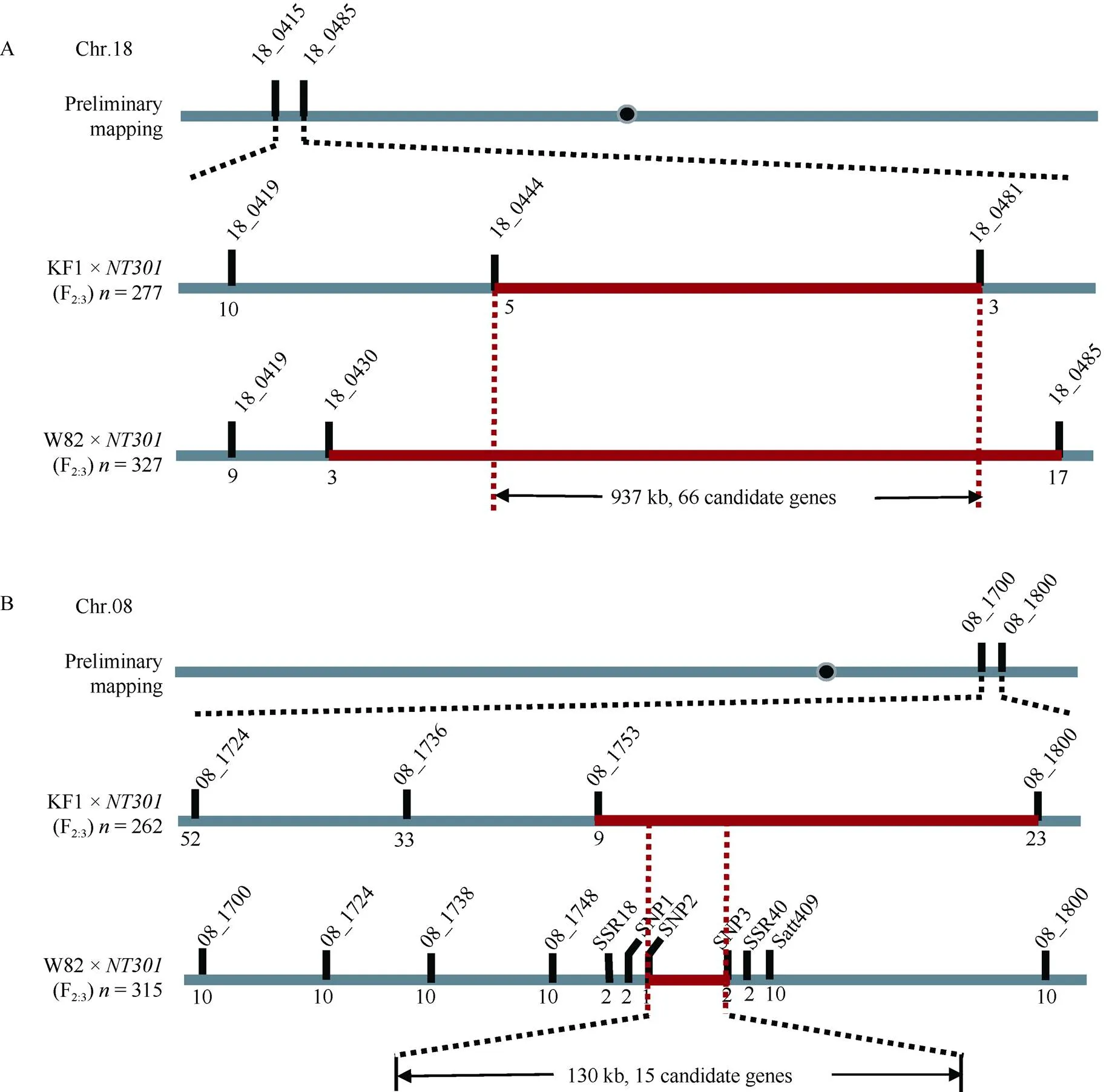
图2 突变体NT301 2对候选基因定位
A: 18号染色体定位; B: 8号染色体精细定位。
A: mapping ofon chromosome 18; B: fine mapping ofon chromosome 8.
整合SoyBase、Phytozome等数据库中的大豆基因组和蛋白组信息, 结合eggNOG-mapper (http:// eggnog-mapper.embl.de/)对差异表达基因进行Pathway注释, 利用KEGG (https://www.genome. jp/kegg/)数据库进行富集分析, 筛选出差异基因显著影响的Pathway (图4)。上调基因所显著参与的合成代谢通路共15个(<0.05, 图4-A), 包括9个代谢通路、5个合成通路和1个植物与病原菌互作通路, 前5条通路达到了极显著水平(0.01)。谷胱甘肽代谢通路基因功能集中在谷胱甘肽转移酶, 如谷胱甘肽S转移酶18/20/22、类γ-谷氨酰转肽酶1; 苯丙素生物合成和苯丙氨酸代谢通路基因功能集中在类过氧化物酶, 如类过氧化物酶4/12/52/73, 有一些涉及病原菌诱导; 缬氨酸、亮氨酸和异亮氨酸降解通路基因功能集中在酰基辅酶A; 氮代谢通路基因功能集中在谷氨酰胺合成酶。下调基因所显著参与的合成代谢通路共有10个(<0.05, 图4-B), 包括8个代谢通路和2个合成通路, 前6条代谢通路达到了极显著水平。卟啉和叶绿素代谢通路基因功能集中在卟啉原脱氨酶、脱羧酶和氧化酶等; 氨基糖和核苷酸糖代谢通路基因功能集中在叶绿体戊糖、己糖激酶、糖苷酶等; 次生代谢产物的生物合成基因功能集中在叶绿体卟啉脱羧酶、转移酶, 糖基合酶、转移酶等, 转移酶、氨酰-tRNA合成基因功能集中在氨酰-tRNA合成酶, 淀粉和蔗糖代谢基因功能集中在叶绿体半乳糖醛酸海藻糖磷酸合酶、果胶酯酶等; 糖酵解/糖异生代谢通路基因功能集中在己糖激酶。
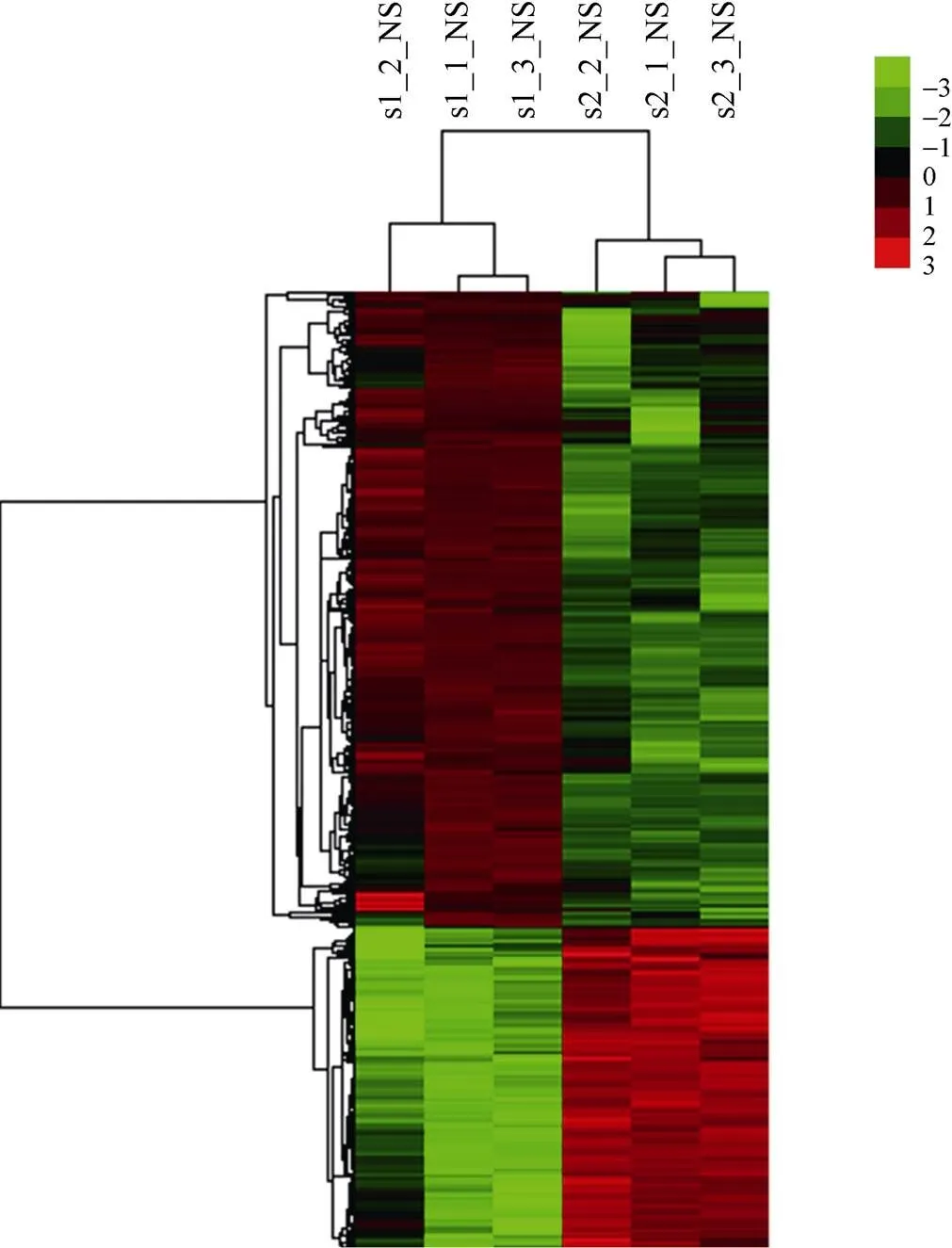
图3 突变体NT301与野生型差异表达基因的聚类图
横坐标代表样品名称及样品的聚类结果, 纵坐标代表差异基因及基因的聚类结果。S1为突变体, S2为野生型, 各3个重复, 不同的行代表不同的基因。颜色代表了基因在样品中的表达量水平。
The horizontal axis represents the sample names and clustering results of the samples, while the vertical axis represents the differentially expressed genes and clustering results of the genes. S1 represents the mutant, and S2 represents the wild type, three times repeat. Different rows represent different genes. Color represents the relative expression level of genes in the samples.
2.4 突变体NT301皱叶候选基因预测
根据生物信息学预测, 18号和8号染色体目标区段内分别有66个和15个候选基因, Phytozome网站上对8号染色体的候选基因功能注释如表3所示。为了进一步预测候选基因, 取V3至V4时期大豆叶片, 提取总RNA, 做半定量和荧光定量PCR, 比较分析候选基因在野生型和突变体叶片中的表达水平。结果如图5所示, 只有基因在突变体与野生型中的差异达到了4倍, 推测基因是突变体的候选基因, 功能注释为UDP糖基转移酶超家族蛋白。将18号和8号染色体定位区间内所有基因的氨基酸序列做了聚类分析, 结果显示8号染色体候选基因与18号染色体聚类在一起, 序列相似度最高, 推测是18号染色体定位区间内的候选基因(附图1)。
在拟南芥中的同源基因是, 编码UDP鼠李糖-花青素-3-葡萄糖苷鼠李糖基转移酶, 是类黄酮合成通路的下游基因, 能够催化花青素苷生成花色素类, 进而在谷胱甘肽转移酶的作用下生成花青素谷胱甘肽。研究认为, 类黄酮基因的表达与生长素极性运输呈负相关, 抑制了生长素转运蛋白在芽和茎中的表达, 从而抑制了腋芽的生长[22]。突变体候选基因的高表达导致下游谷胱甘肽也随之升高, 这与表达谱芯片的结果一致, 由此影响了上游类黄酮基因的表达, 使生长素运输受到影响, 最终导致叶脉与叶肉细胞发育不协调, 形成皱叶的表型。

表1 杂交组合F2和F2:3群体野生型和突变体植株分离率适合性检验
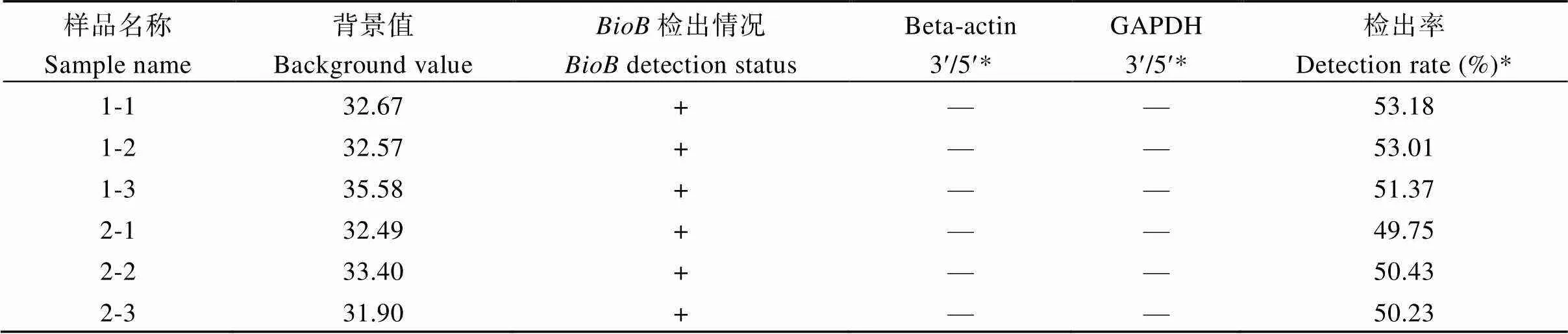
表2 芯片试验质控情况
Beta-actin 3¢/5¢*与GAPDH 3¢/5¢*: 管家基因3¢端信号与5¢端信号比值至少有一个不大于3, 此标准仅适用于以下8种表达谱芯片: 人、小鼠、大鼠、斑马鱼、线虫、果蝇、酵母和大肠杆菌表达谱芯片。检出率Detection rate (%)*: 检出点总数与全部探针数的比值为该芯片的检出率。
Beta-actin 3¢/5¢* and GAPDH 3¢/5¢*: the ratio of the 3' ends signal to the 5' ends signal of the housekeeping genes should not exceed 3. This standard only applies to the following 8 expression profile chips: human, mouse, rat, zebrafish, nematode, fruit fly, yeast, andexpression profile chips. Detection rate (%)*: the ratio of the total number of detected probes to the total number of probes is the detection rate of the chip.

表3 rl2位点候选基因功能注释

(图4)
A: 上调基因富集的KEGG通路; B: 下调基因富集的KEGG通路。图中–log10越大, 表示该通路越显著。横坐标表示富集值, 纵坐标表示富集的通路。圆圈大小表示基因数量。
A: the enrichment of upregulated genes in the KEGG; B: the enrichment of downregulated genes in the KEGG. The figure shows that the –log10is greater, the pathway is more significant. The horizontal axis represents the enrichment value, and the vertical axis represents the pathway of enrichment. The size of the circle indicates the number of genes.

图5 8号染色体定位区间内基因的表达分析
A: 8号染色体定位区间内基因半定量PCR分析; B: 8号染色体定位区间内基因荧光定量PCR分析。红色方框表示野生型和突变体表达量差异最大的基因, 采用学生测验进行显著性检验(**:≤ 0.01), 误差线表示标准差(= 3)。
A: the semi quantitative PCR analysis of genes in the mapping interval of chromosome 8; B: the quantitative RT-PCR analysis of genes in the mapping interval of chromosome 8. The red box indicates the most differentially expressed gene between wild type and mutant. The significant test was carried out by the student’s-test (**:≤ 0.01). The error bars indicate the SDs (= 3).
3 讨论
叶片皱缩受2对隐性基因的控制, 分别位于18号和8号染色体上, 只有2对基因同时突变为隐性时, 才出现类似花叶病毒感染的特征, 其中任何一对基因为显性时, 都不出现花叶病毒的症状, 所以推测这2对基因是同源基因且功能冗余。根据SoyBase网站参考基因组的序列信息, 18号染色体定位区间内有两段序列的重复序列刚好位于8号染色体的定位区间之内, 且此区域内8号染色体的基因密度大于18号染色体。根据前人的研究结果, 由于不对称进化, 18号染色体着丝粒及周围区域与8号染色体短臂有大量的同源基因, 全基因组复制事件之后, 这些基因经历了从常色状态到异色状态的完全或部分切换, 或者刚好相反[23], 因此推测和基因也是由同源基因进化而来的。
在大豆基因组测序没有完成之前, 研究者利用经典遗传学中同源基因的遗传特性及基因定位能够推断其同源连锁群, 这对于研究大豆的进化是十分有利的。Lohnes等[24-25]揭示了和位点共同调控子叶颜色的遗传机制, 只有2个位点都为隐性即时, 子叶为绿色, 其余基因型都为黄色, 并且验证了和所在连锁群为部分同源连锁群。Fang等[26]进一步研究发现,和与拟南芥和水稻中的SGR (STAY-GREEN)基因是同源基因, 与本研究不同的是,与在衰老早期均抑制了叶绿素的降解, 但在衰老后期的抑制作用没有强烈, 表明基因与基因的功能并非完全冗余。
为适应进化, 多倍体植物中重复基因可能发生假基因化、亚功能化或新功能化[27], Ji等[28]认为染色体或片段重复在基因家族的扩张过程中起到了重要作用。重复基因往往在转录水平上有差异, 从而引起表型的变化[29]。大豆生长习性基因()有4个同源基因, 但功能已经发生了变化, 它们在不同发育阶段有不同的表达模式, 而且在群体中的变异位点也是不同的[30]。多倍体由于增加了遗传变异和复制基因的缓冲作用, 对极端环境具有更强的耐受性, Chen等[31]筛选到一个新的既受到除草剂选择也发生了多倍化扩增的基因, 并验证了该基因与氰氟草酯和恶唑酰草胺的结构相互作用, 从分子水平揭示了多倍体杂草具有更强的除草剂适应性的原因。
到目前为止, 大豆中对于类病变皱叶突变体相关基因挖掘较少, 其所在代谢通路上下游关系还不明确。而拟南芥中研究较多, 如过表达或者下调其靶基因, 影响茉莉酸合成通路, 促进细胞增殖或者叶片皱缩[32]。拟南芥胆色素原脱氨酶缺失导致叶片发育畸形, 细胞坏死, 生长受到抑制, 开花延迟, 并触发植物防御机制激活[33]。拟南芥突变体叶脉扭曲并开叉形成双脉, 并且侧根异常弯曲, 细胞非正常扩张, 游离生长素水平正常, 但是运输受到损害[34]。拟南芥和突变体叶脉不能形成网状结构, 变成沿主脉的狭长一条, 且形状不规则, 叶缘有缺刻, 作者推测是由于生长素分布不均匀导致的[35]。叶脉与叶肉发育不协调, 叶缘形成不规则褶皱, 叶面积大大减小, 而叶肉细胞密度大大增加, 推测可能跟生长素分配不均有关。
4 结论
本研究利用大豆类病变皱叶突变体与W82、KF1、KF35构建的F2、F2:3杂交群体, 用SSR和SNP等分子标记进行基因定位, 将目的基因1 ()缩小到18号染色体937 kb区间内, 包含66个候选基因, 将目的基因2 ()缩小到8号染色体130 kb区间内, 包含15个候选基因, 并根据表达谱芯片和qPCR的结果, 推测是候选基因。
[1] 吕慧颖, 王道文, 葛毅强, 魏珣, 邓向东, 杨维才, 田志喜. 大豆育种行业创新动态. 植物遗传资源学报, 2018, 19: 464–467. Lyu H Y, Wang D W, Ge Y Q, Wei X, Deng X D, Yang W C, Tian Z X. Innovation of soybean breeding industry.,2018, 19: 464–467 (in Chinese with English abstract).
[2] 田志喜, 刘宝辉, 杨艳萍, 李明, 姚远, 任小波, 薛勇彪. 我国大豆分子设计育种成果与展望. 中国科学院院刊, 2018, 33: 915–922. Tian Z X, Liu B H, Yang Y P, Li M, Yao Y, Ren X B, Xue Y B. Update and prospect of soybean molecular module-based designer breeding in China.,2018, 33: 915–922 (in Chinese with English abstract).
[3] Huang Q N, Yang Y, Shi Y F, Chen J, Wu J L. Spotted-leaf mutants of rice ()., 2010, 17: 247–256.
[4] Wu C J, Bordeos A, Madamba M R S, Baraoidan M, Ramos M, Wang G L, Leach J E, Leung H. Rice lesion mimic mutants with enhanced resistance to diseases., 2008, 279: 605–619.
[5] Gray J, Close P S, Briggs S P, Johal G S. A novel suppressor of cell death in plants encoded by thegene of maize., 1997, 89: 25–31.
[6] Dietrich R A, Richberg M H, Schmidt R, Gean C, Dangl J L. A novel zinc finger protein is encoded by thegene and functions as a negative regulator of plant cell death., 1997, 88: 685–694.
[7] Tang X, Xie M, Kim Y J, Zhou J, Klessig D F, Martin G B. Overexpression ofactivates defense responses and confers broad resistance., 1999, 11: 15–29.
[8] Yamanouchi U, Yano M, Lin H, Yamada K. A rice spotted leaf gene,, encodes a heat stress transcription factor protein., 2002, 99: 7530–7535.
[9] Wang Y, Liu M, Ge D, Bhat J A, Li Y, Kong J, Liu K, Zhao T. Hydroperoxide lyase modulates defense response and confers lesion-mimic leaf phenotype in soybean ((L.) Merr.)., 2020, 104: 1315–1333.
[10] Ma J, Yang S, Wang D, Tang K, Feng X. Genetic mapping of a light-dependent lesion mimic mutant reveals the function of coproporphyrinogen iii oxidase homolog in soybean., 2020, 11: e557.
[11] Stephens P A, Barwale U B, Nickell C D, Widholm J M. A cytoplasmically inherited, wrinkled-leaf mutant in soybean., 1991, 82: 71–73.
[12] Wilcox J R, Abney T S. Inheritance of a narrow, rugose-leaf mutant in., 1991, 82: 421–423.
[13] 聂智星, 代金英, 吉家正, 陈薇, 赵团结. 大豆叶突变体的发掘与特性分析. 江苏农业科学, 2013, 41(1): 86–88.Nie Z X, Dai J Y, Ji J Z, Chen W, Zhao T J. Identification and characterization of soybean leaf mutant.,2013, 41(1): 86–88 (in Chinese).
[14] Singh S P, Molina A. Inheritance of crippled trifoliolate leaves occurring in interracial crosses of common bean and its relationship with hybrid dwarfism., 1996, 87: 464–469.
[15] Song X, Wei H, Cheng W, Yang S, Zhao Y, Li X, Luo D, Zhang H, Feng X. Development of INDEL markers for genetic mapping based on whole genome resequencing in soybean., 2015, 5: 2793–2799.
[16] Ochar K, Bo-Hong S U, Zhou M M, Liu Z X, Gao H W, Flamlom S, Qiu L J. Identification of the genetic locus associated with the crinkled leaf phenotype in a soybean (L.) mutant by BSA-Seq technology., 2022, 21: 3524–3539.
[17] 孙红正, 葛颂. 重复基因的进化: 回顾与进展. 植物学报, 2010, 45: 13–22. Sun H Z, Ge S. Review the evolution of duplicate genes., 2010, 45: 13–22 (in Chinese with English abstract).
[18] Schmutz J, Cannon S B, Schlueter J, Ma J, Jackson S A. Genome sequence of the palaeopolyploid soybean., 2010, 463: 178–183.
[19] Wang Y, Chen W, Zhang Y, Liu M, Kong J, Yu Z, Jaffer A M, Gai J, Zhao T. Identification of two duplicated loci controlling a disease-like rugose leaf phenotype in soybean., 2016, 56: 1611–1618.
[20] Song Q J, Jia G F, Zhu Y L, Grant D, Nelson R T, Hwang E Y, Hyten D L, Cregan P B. Abundance of SSR motifs and development of candidate polymorphic SSR markers (BARCSOYSSR_ 1.0) in soybean., 2010, 50: 1950–1960.
[21] 李强, 万建民. SSRHunter, 一个本地化的SSR位点搜索软件的开发. 遗传, 2005, 27: 808–810. Li Q, Wan J M. SSRHunter, development of a local searching software for SSR sites., 2005, 27: 808–810 (in Chinese with English abstract).
[22] Lazar G, Goodman H M., a regulator of the flavonoid pathway, controls vegetative axillary bud outgrowth in., 2006, 103: 472–476.
[23] Du J, Tian Z, Sui Y, Zhao M, Song Q, Cannon S B, Cregan P, Ma J. Pericentromeric effects shape the patterns of divergence, retention, and expression of duplicated genes in the paleopolyploid soybean., 2012, 24: 21–32.
[24] Lohnes D G, Specht J E, Cregan P B. Evidence for homoeologous linkage groups in the soybean., 1997, 37: 254–257.
[25] Chao W S, Liu V, Thomson W W, Platt K, Walling L L. The impact of chlorophyll-retention mutations,and, during embryogeny in soybean., 1995, 107: 253–262.
[26] Fang C, Li C, Li W, Wang Z, Zhou Z, Shen Y, Tian Z. Concerted evolution ofandto regulate chlorophyll degradation in soybean., 2014, 77: 700–712.
[27] Innan H, Kondrashov F. The evolution of gene duplications: classifying and distinguishing between models., 2010, 11: 97–108.
[28] Ji Q, Zhang L S, Wang Y F, Wang J. Genome-wide analysis of basic leucine zipper transcription factor families in,and., 2009, 13: 174–182.
[29] Wang Y P, Wang X Y, Paterson A H. Genome and gene duplications and gene expression divergence: a view from plants., 2012, 1256: 1–14.
[30] Tian Z X, Wang X, Lee R, Li Y, Specht J E, Nelson R L, McClean P E, Qiu L, Ma J. Artificial selection for determinate growth habitin soybean., 2010, 107: 8563–8568.
[31] Chen K, Yang H, Peng Y, Liu D, Zhang J, Zhao Z, Wu L, Lin T, Bai L. Genomic analyses provide insights into the polyploidization-driven herbicide adaptation inweeds., 2023, 21: 1642–1658.
[32] Schommer C, Palatnik J F, Aggarwal P, Chetelat A, Cubas A, Farmer E E, Nath U, Weigel D. Control of jasmonate biosynthesis and senescence bytargets., 2008, 6: e230.
[33] Quesada V, Sarmiento-Mañús R, González-Bayón R, Hricová A, Ponce M R, Micol J L. PORPHOBILINOGEN DEAMINASE deficiency alters vegetative and reproductive development and causes lesions in., 2013, 8: e53378.
[34] Carland F M, McHale N A.: a gene involved in auxin transport and vascular patterning in., 1996, 122: 1811–1819.
[35] Cnops G, Neyt P, Raes J, Petrarulo M, Nelissen H, Malenica N, Luschnig C, Tietz O, Ditengou F, Palme K, Azmi A, Prinsen E, Lijsebettensa M V. Theandgenes function in several patterning processes during early leaf development in., 2006, 18: 852–866.
附表1 本研究所用到的引物
Table S1 Primers used in this study
引物名称Primer name正向引物Forward primer (5¢-3¢)反向引物Reverse primer (5¢-3¢) BARCSOYSSR_18_0415GCTTGGCGAATTCCATCTAAATTCATTTCACATCCCAGGC BARCSOYSSR_18_0419TTTATTATGGGGATCAAATTAAAACTTCGGAGTTAGCCAAATCAAAT BARCSOYSSR_18_0485AAGGATTGGCAAAGCGATTAAAAAACAGGGTTTGGTGGGT BARCSOYSSR_18_0444TTGACGGCCTTATTTTGGACGGTGGCTGAATCCAAGACAT BARCSOYSSR_18_0481ATCTCAAGGATCTGGCAAGCAATATTCTTTGGCGGTGGTG BARCSOYSSR_08_1700CCTTTAATCAACTCTGTGAGATCGGCATAATGCTATTCTCCGCA BARCSOYSSR_08_1800ACAAGGAGATTTGGCTTTGCCCGGAGATCGATAAGTTGCT BARCSOYSSR_08_1724CATTGGAGCTTGGTAAGGGAGGGAGCAGAGAACTTGAGCA BARCSOYSSR_08_1736TCCTTTTTGAAAACGAAATTCAGAGTACATTGGAATAACTGTGCAA BARCSOYSSR_08_1724CATTGGAGCTTGGTAAGGGAGGGAGCAGAGAACTTGAGCA BARCSOYSSR_08_1738TGTTAGGGACACACTCAACCCCCACGAGATAAGACGAGCAA BARCSOYSSR_08_1753CCAGCCACTTCACAGACTGATTGGGTTATCTGTTTGCTTTGTT SSR18GGCTTGACTCTCTGAATCTGTTTAAACAACTTTGAGCCAACGGC SSR40ACAAACCTGGTTCGGCTATGTTTACCAAAGCAGTGGGAGCC Satt409CCTTAGACCATGAATGTCTCGAAGATACTTAAGGACACGTGGAAGATGACTAC SNP1TTCGAAGGAACTTACTCATTACCAACATTCTCAAGCCCAAGG SNP2AGCACGATTCTACCTCCGAATGGTAGGCAATCTGAACTCATCA SNP3AGGGATATGGTTCATCTTTCATCTATGATGTGTTATGTGCATAT Glyma.08g332000QRTGGCGTCCAGGATTTACAAAGTTGATATTGCACCAGGCTTTGA Glyma.08g332100QRTCATGGCTGGCTTTAAGAGGAAGTATCAAAGAAGGAGCCGTT Glyma.08g332200QRTACTACGGTGCATCAGAGATTCTCAGTGCAATCATAACTGTGGTA Glyma.08g332300QRTGGCCGCTACCATCAACTTCAAGCTCTAGTGAACTACGGCATT Glyma.08g332400QRTTGCCTTCACACATTATAAACAGGTATGGTTTCCACTTCCGACCC Glyma.08g332500QRTAATGAGAGAACAAAGGGAAGAGGGAGTCACAAAGCAACCCACAG Glyma.08g332600QRTGATGTGGTGCAGAGCAAACCCACCATCGTCAAGTTGTGGC Glyma.08g332700QRTTGGTGATGAAGAACAACAGCACAAGAAGTCCTGGCCTAGC Glyma.08g332800QRTTGCTGAATCTGGCATGAACCGACGGATGGCGCAAAACAAG Glyma.08g332900QRTTTGTTGACAGTGGTATTGGCAAACCCCACACCAAACTGTCC Glyma.08g333000QRTCGGTTTGTCTATATTGTTGAAGGTTTCTCCACTGAACTGGTCCAC Glyma.08g333100QRTCCGCCCAAACCTCTGAAGATCAGACTGGAAGTTGCCAGGTA Glyma.08g333200QRTCAAGAACTGCAACTGAAAATGGTTGCATGCCAAAACTGTACACATCAC Glyma.08g333300QRTCGCCAGAACTTTGCAAGGAAACCCGAACCATTCACGACTT Glyma.08g333400QRTTGAAGCATGGCGAAGCAGTACGAACACGTGGGTCATGGAA GmActinGGTGGTTCTATCTTGGCATCCTTCGCTTCAATAACCCTA
附图1 突变体18号和8号染色体定位区间内候选基因氨基酸序列聚类分析
Fig. S1 Clustering analysis of amino acid sequences of candidate genes within the mapping intervals on chromosomes 18 and 8 in the mutant
红色方框表示8号染色体表达量差异最大的基因和18号染色体的同源基因。
The red box indicates the most differentially expressed geneon chromosome 8 and its homologous geneon chromosome 18.
Genetic analysis and two pairs of genes mapping in soybean mutantwith disease-like rugose leaf
WANG Ya-Qi1, XU Hai-Feng1, LI Shu-Guang1, FU Meng-Meng1, YU Xi-Wen1, ZHAO Zhi-Xin1, YANG Jia-Yin1,*, and ZHAO Tuan-Jie2,*
1Huaiyin Institute of Agricultural Sciences of Xuhuai Region in Jiangsu / Huai’an Key Laboratory for Agricultural Biotechnology / Key Laboratory of Germplasm Innovation in Lower Reaches of the Huaihe River, Ministry of Agriculture and Rural Affairs, Huai’an 223001, Jiangsu, China;2Soybean Research Institute, Nanjing Agricultural University / National Center for Soybean Improvement (Nanjing) / Key Laboratory for Biology and Genetic Improvement of Soybean (General), Ministry of Agriculture and Rural Affairs / National Key Laboratory of Crop Genetics and Germplasm Enhancement, Nanjing 210095, Jiangsu, China
Research on lesion mimic mutant, mining resistance genes, and developing superior disease-resistant new soybean varieties by molecular design breeding methods can contribute to the alleviating the environmental pollution caused by chemical pesticides and drug resistance to disease. In this study, the disease-like rugose leaf mutantobtained by60Coγ mutagenesis as the male parent was crossed with W82, KF1, and KF35, respectively, to construct F2and F2:3segregating populations. Using SSR and SNP markers, target gene 1 () was narrowed to 937 kb on chromosome 18 with 66 genes and target gene 2 () was narrowed to 130 kb on chromosome 8 with 15 genes. The gene expression patterns of the wild type andwere compared using gene chip technology, and the KEGG pathways of the differentially expressed genes were assessed. Moreover, semi quantitative and quantitative RT-PCR methods were used to analyze the relative expression levels of candidate genes on chromosome 8. The results showed that the relative expression level ofinwas four times higher than the wild type. In contrast, the expression levels of other genes showed no more than double difference. Therefore, we suggest thatmay be a candidate gene for.
soybean; disease-like mutant; rugose leaf; gene expression profiles
10.3724/SP.J.1006.2024.34106
本研究由国家自然科学基金项目(32201729), 淮安市自然科学研究计划项目(联合专项, HABL202120), 淮安市农业科学研究院高层次引进人才科研启动发展基金(0112023014B), 淮安市农业科学研究院科研发展基金(HNY202221), 江苏省种业振兴揭榜挂帅项目(JBGS[2021]057)和江苏省现代作物生产协同创新中心项目(JCIC-MCP)资助。
The study was supported by the National Natural Science Foundation of China (32201729), the Natural Science Research Program of Huai’an (Joint Special Project, HABL202120), the Scientific Research Fund of Startup and Development for Introduced High-level Talents, Huai’an Academy of Agricultural Sciences (0112023014B), the Research and Development Fund Project of Huai’an Academy of Agricultural Sciences (HNY202221), the Core Technology Development for Breeding Program of Jiangsu Province (JBGS[2021]057), and the Jiangsu Collaborative Innovation Center for Modern Crop Production (JCIC-MCP) Program.
赵团结, E-mail: tjzhao@njau.edu.cn; 杨加银, E-mail: hynksyjy@163.com
E-mail: yqwang_01@126.com
2023-06-27;
2023-10-23;
2023-11-14.
URL: https://link.cnki.net/urlid/11.1809.S.20231113.1429.005
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
猜你喜欢
安徽农学通报(2022年6期)2022-04-07 21:30:29
科学之谜(2019年3期)2019-03-28 10:29:44
科学之谜(2018年8期)2018-09-29 11:06:46
畜牧与饲料科学(2018年3期)2018-05-08 02:48:35
湖南林业科技(2017年1期)2017-02-06 05:28:55
安徽医科大学学报(2016年12期)2017-01-15 14:21:44
山东农业工程学院学报(2016年6期)2016-12-01 05:38:19
恋爱婚姻家庭·养生版(2016年9期)2016-09-07 11:25:01
中央民族大学学报(自然科学版)(2015年2期)2015-06-09 08:45:16
山东医药(2015年40期)2015-02-28 14:28:45
