
高效液相色谱法测定醋酸氟卡尼片的含量
2024-03-13 07:49徐硕邝咏梅徐文峰金鹏飞北京医院药学部国家老年医学中心中国医学科学院老年医学研究院北京市药物临床风险与个体化应用评价重点实验室北京医院北京100730
中南药学 2024年1期
徐硕,邝咏梅,徐文峰,金鹏飞(北京医院药学部,国家老年医学中心,中国医学科学院老年医学研究院,北京市药物临床风险与个体化应用评价重点实验室(北京医院),北京 100730)
氟卡尼是钠通道阻滞药,属于Ⅰc类抗心律失常药。醋酸氟卡尼片主要用于治疗室性心律失常,包括室性期前收缩及室性心动过速、室上性期前收缩、室上性心动过速、心房扑动、心房颤动、预激综合征合并室上性心动过速等[1-4]。醋酸氟卡尼片收载于《美国药典》[5]和《日本药典》[6]。目前,该药还没有在我国上市,也未在《中国药典》中收载。关于醋酸氟卡尼片的研究主要集中在药理作用,其含量测定方面的研究报道非常有限,仅有一篇早期的报道[7],采用HPLC法和紫外分光光度法进行定量。利用HPLC法是通过内标法测定,溶液配制操作复杂。文献报道以及《日本药典》收载的以紫外分光光度法测定醋酸氟卡尼含量,其专属性相对较差,《美国药典》中是采用HPLC法,流动相为高浓度缓冲盐溶液,梯度洗脱,该方法于2019年进行修订并沿用至今。而在此之前《美国药典》[8]中收录的方法为采用C8分析柱,并且用四相混合溶液的复杂流动相体系,不利于色谱柱和液相系统的长时间运行。因此,有必要建立一种普适性强并且快速简便的方法,用于醋酸氟卡尼片中醋酸氟卡尼的定量分析。本研究通过HPLC技术,建立了醋酸氟卡尼片的含量测定方法,该方法简便、快速、准确,可为该药品的质量评价提供科学依据,现报道如下。
1 仪器与试药
1.1 仪器
高效液相色谱仪 [Agilent 1260型,包括四元梯度泵(G1311C),自动进样器(G1329B),柱温箱(G1316A),二极管阵列检测器(DAD)(G4212B),色谱工作站(LC OpenLAB),美国Agilent Technologies有限公司];电子分析天平(XP-205型,精度:十万分之一,瑞士Mettler Toledo集团);SPX-250 IC微电脑人工气候箱(上海博迅实业有限公司);数控超声波清洗器(KQ-300DB型,昆山市超声仪器有限公司)。
1.2 试药
乙腈、磷酸(Thermo Fisher Scientific有限公司产品,色谱纯,批号分别为192855、158625);磷酸二氢铵、冰醋酸、盐酸、30%过氧化氢(国药集团化学试剂有限公司产品,分析纯),氢氧化钠(北京化工厂,分析纯);水为二级实验用水。醋酸氟卡尼对照品 [批号:G0K124,纯度:99.7%,USP];醋酸氟卡尼片(规格:含醋酸氟卡尼100 mg/片,批号:GUL050A、GUD052B、GTB072E,英国Inova Pharmaceuticals公司)。
2 方法与结果
2.1 色谱条件
色谱柱采用Alltima C18柱(150 mm×4.6 mm,5 µm);流动相为乙腈-0.02 mol·L-1磷酸二氢铵溶液(含0.2%三乙胺)(用磷酸调至pH 3.0)(35∶65);流速为1.0 mL·min-1;检测波长为300 nm;柱温为30℃;进样量为10 µL。
2.2 对照品溶液的配制
精密称取醋酸氟卡尼对照品适量,置于500 mL量瓶中,加入2%冰醋酸溶液40 mL,超声使其溶解,再加入流动相稀释至刻度,即得质量浓度约为2 mg·mL-1的对照品储备液。分别精密量取对照品储备液2.5、4、5、6、7.5 mL,置于25 mL量瓶中,加流动相稀释至刻度,摇匀,即得5个不同浓度的线性系列溶液(质量浓度分别为196.0、313.6、392、470.4、588.0 μg·mL-1)。将中间浓度的溶液(392 μg·mL-1)作为标准工作液,用作方法的系统适用性考察。
2.3 供试品溶液的配制
取醋酸氟卡尼片20片,精密称定,将其置于乳钵中研细,精密称取约相当于醋酸氟卡尼100 mg的样品(约为0.26 g),置于250 mL量瓶中,加入2%冰醋酸溶液20 mL,超声提取(功率为300 W,频率为40 kHz)30 min,再加流动相定容至刻度,摇匀,经0.22 µm微孔滤膜滤过,取续滤液,即为供试品溶液。
2.4 阴性样品溶液的配制
依据醋酸氟卡尼片药品说明书提供的配方制得的不含醋酸氟卡尼的粉末作为阴性样品。精密称定该粉末适量,按照“2.3”项下方法操作,即得阴性样品溶液。
2.5 系统适用性、线性关系考察、检测限和定量限
在“2.1”项色谱条件下,将标准工作液连续进样6次,醋酸氟卡尼色谱峰的平均理论塔板数为(4839.3±10.3)(n=6)。精密吸取“2.2”项下5个不同浓度的对照品溶液各10 µL,在“2.1”项色谱条件下进样测定,记录醋酸氟卡尼峰面积,以该成分的质量浓度(μg·mL-1)为横坐标(X),峰面积为纵坐标(Y),进行回归分析,得到回归方程为:Y=3.549X+3.566,r=1.000,醋酸氟卡尼在196.0~588.0 μg·mL-1与峰面积呈良好的线性关系。
取质量浓度为196.0 μg·mL-1的对照品溶液,加入流动相进行稀释,进样测定,以低浓度对照品信噪比约为3.0和10.0时的浓度分别作为检测限(LOD)和定量限(LOQ),结果醋酸氟卡尼的LOD为0.061 µg·mL-1,LOQ为0.196 µg·mL-1。
2.6 专属性试验
2.6.1 基质干扰试验 精密吸取上述阴性样品溶液、醋酸氟卡尼对照品溶液以及供试品溶液各10µL,进样分析,按照“2.1”项下色谱条件检测,结果显示样品中的辅料不干扰醋酸氟卡尼的测定(见图1)。

图1 阴性样品溶液(A)、对照品溶液(B)和供试品溶液(C)的HPLC色谱图Fig 1 HPLC chromatograms of negative sample solution(A),reference substance solution(B)and sample solution(C)
2.6.2 强制降解试验 为进一步考察方法的专属性,对供试品进行强制降解试验,参照国际通行的人用药品注册技术要求国际协调会(ICH)方法学验证原则进行[9-10]。考察的条件包括酸化、碱化、光照、氧化和高温。取醋酸氟卡尼片(批号:GUL050A)样品粉末,精密称定5份,每份约为0.26 g(约相当于醋酸氟卡尼100 mg),置于250 mL量瓶中,进行处理如下:
① 酸降解:分别配制浓度为1 mol·L-1的盐酸溶液和1 mol·L-1的氢氧化钠溶液,量取1 mol·L-1的盐酸溶液2 mL加入样品中,置于室温条件下放置6 h后,加入1 mol·L-1的氢氧化钠溶液,使其中和至中性,依照“2.3”项下方法操作,制得供试品溶液。② 碱降解:量取2 mL 1 mol·L-1的氢氧化钠溶液加入样品中,置于室温条件下放置6 h后,加入1 mol·L-1的盐酸溶液,使其中和至中性,按照“2.3”项下方法操作,制得供试品溶液。③ 光降解:将样品放入微电脑人工气候箱中,光照强度设置为10 000 lx,在该条件下照射6 h,依照“2.3”项下方法操作,制得供试品溶液。④ 氧化降解:在样品中加30%过氧化氢溶液2 mL,室温下放置6 h,依照“2.3”项下方法操作,制得供试品溶液。⑤ 高温降解:将样品放入烘箱中,80℃加热6 h,取出,待其冷却到室温后,依照“2.3”项下方法操作,制得供试品溶液。
将上述溶液各进样10 µL,在“2.1”项色谱条件下进行检测。结果显示:与未经过强制降解试验的样品比较,醋酸氟卡尼在高温条件较为稳定,未发生降解;在光照、碱化、酸化及氧化条件下有所降解,色谱峰峰面积略有降低,氧化条件下降解较为明显,可见降解产物色谱峰,但对醋酸氟卡尼的定量分析不会造成干扰(见图2)。
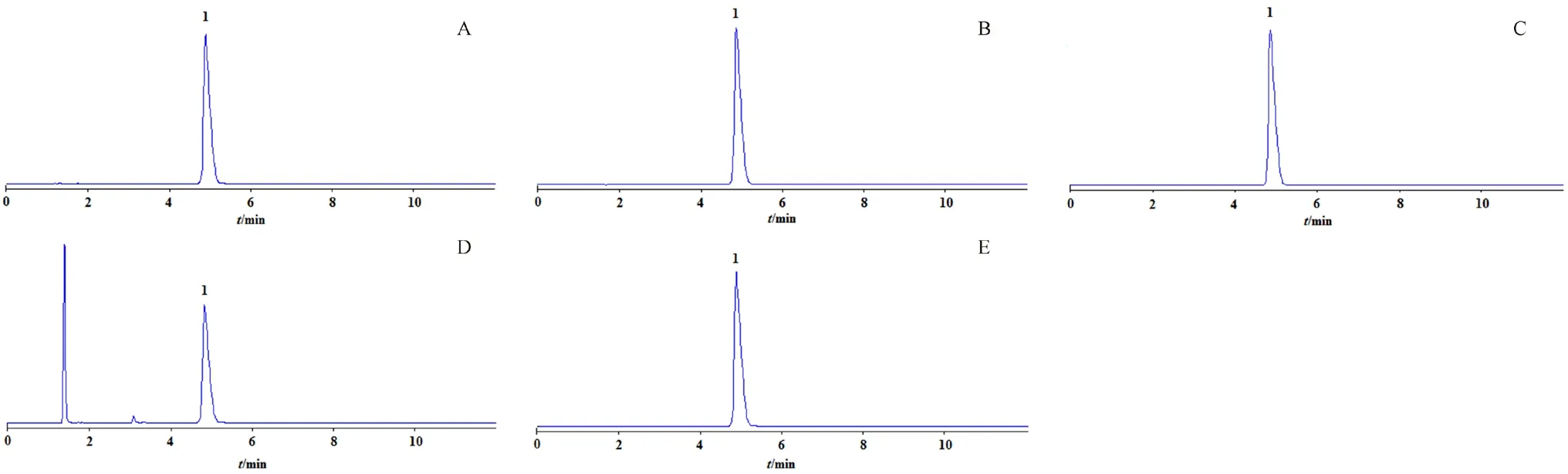
图2 醋酸氟卡尼片样品强制降解试验HPLC色谱图Fig 2 HPLC chromatogram of forced degradation test for flecainide acetate tablets samples
2.7 精密度试验
取批号为GUL050A的醋酸氟卡尼片制得的供试品溶液,连续进样6针,记录峰面积,结果醋酸氟卡尼色谱峰峰面积的RSD值为0.14%(n=6),表明精密度良好。
2.8 重复性试验
精密称取批号为GUL050A的醋酸氟卡尼片样品6份,每份约为0.26 g,按照“2.3”项下方法操作,制得供试品溶液,进样分析,以外标法计算含量,结果醋酸氟卡尼的平均含量为98.15%,RSD值为0.53%(n=6),表明该方法具有良好的重复性。
2.9 稳定性试验
取批号为GUL050A的供试品溶液,置于室温条件下,分别在配制后的0、2、4、8、12、24 h进样检测,记录峰面积值,计算醋酸氟卡尼色谱峰峰面积的RSD值为0.49%(n=6),表明供试品溶液在24 h内具有良好的稳定性。
2.10 加样回收试验
取批号为GUL050A的样品,研细,精密称取粉末9份,每份约为0.13 g,分别精密加入“2.2”项下对照品储备液20、25、30 mL,按照“2.3”项下方法制备,每个浓度制备3份样品,进样测定,计算回收率和RSD,结果如表1所示。

表1 回收试验结果Tab 1 Recovery test
2.11 耐用性考察
分别采用Alltima C18柱(150 mm×4.6 mm,5 µm)、Shimadzu C18柱(150 mm×4.6 mm,5µm)、Waters C18柱(150 mm×4.6 mm,5 µm)进行试验,结果显示不同品牌色谱柱对醋酸氟卡尼的保留时间、峰形无影响。改变色谱条件中的柱温(30±2)℃,结果显示醋酸氟卡尼保留时间基本不变,RSD为0.63%。改变色谱条件中的流速[(1.0±0.2)mL·min-1],醋酸氟卡尼的保留时间分别为4.0、4.9、6.1 min。结果表明本方法的HPLC条件耐用性良好,色谱参数的微调不会影响检测结果。
2.12 样品含量测定
采用已建立的方法测定了3批醋酸氟卡尼片,每批样品精密称取2份,按照“2.3”项下方法制得供试品溶液,进样分析,计算醋酸氟卡尼含量,取平均值作为含量测定结果,见表2。

表2 样品中醋酸氟卡尼的含量Tab 2 Content of flecainide acetate in samples
3 讨论
3.1 检测波长的选择
设定醋酸氟卡尼的检测波长是根据其在流动相中的紫外吸收谱图,谱图显示该成分的最大吸收为204 nm和300 nm,将300 nm选定为含量测定的检测波长。
3.2 色谱条件的优化
考察不同流动相体系[乙腈-0.1 mol·L-1磷酸二氢钠溶液(用磷酸调至pH 3.0)、乙腈-0.05 mol·L-1醋酸铵溶液(用冰醋酸调至pH 3.0)、乙腈-0.05 mol·L-1磷酸二氢铵溶液(含0.1%辛烷磺酸钠)(用磷酸调至pH 3.0)、乙腈-0.02 mol·L-1磷酸二氢铵溶液(用磷酸调至pH 3.0)、乙腈-0.02 mol·L-1磷酸二氢铵溶液(含0.2%三乙胺)(用磷酸调至pH 3.0)]对醋酸氟卡尼色谱峰的影响,结果表明将乙腈-0.02 mol·L-1磷酸二氢铵溶液(含0.2%三乙胺)(用磷酸调至pH 3.0)作为流动相时待测成分峰形较好,理论塔板数大于4500,将其选定为流动相。考察了以乙腈-0.02 mol·L-1磷酸二氢铵溶液(含0.2%三乙胺)(用磷酸调至pH 3.0)作为流动相,调整有机相和水相不同比例,以及设定不同流速(0.5、0.8、1.0 mL·min-1)对待测成分保留时间的影响,结果显示当乙腈和水相的比例为35∶65,流速采用1.0 mL·min-1时,醋酸氟卡尼的保留时间约为5 min,较为合适。
3.3 提取溶剂的选择
在含量测定分析中选择常用的超声提取法作为样品的提取方法,比较了以乙腈、甲醇、2%冰醋酸溶液、0.2%冰醋酸溶液作为提取溶剂的影响,结果表明用2%冰醋酸溶液20 mL作为提取溶剂能够提取完全,之后再加流动相稀释至刻度能使色谱峰峰形良好。
3.4 样品取样量的选择
制备供试品溶液时,对称取不同样品粉末量(约相当于醋酸氟卡尼25、100、250 mg)进行比较,结果显示当称取量约为相当于醋酸氟卡尼100 mg,加2%冰醋酸溶液20 mL,再用流动相稀释至250 mL时目标测定成分具有合适的响应值。
3.5 测定方法的改进
现行版《日本药典》[6]以及文献报道[7]的醋酸氟卡尼片含量测定方法为采用紫外分光光度法,仪器设备和操作比较简单,但其专属性不如HPLC法。文献中是通过吸光系数法进行定量,未配制对照品溶液,吸光系数法有可能造成测定结果的不准确,如果杂质或辅料在选定的测定波长处有吸收,则会导致结果偏高。HPLC法具有定量准确、分离性能高、专属性强等特点,在药品定量分析中具有一定优势[11]。《美国药典》[5]中收载的醋酸氟卡尼片,是通过HPLC法测定其含量,流动相为乙腈-100 mmol·L-1乙酸铵溶液(用乙酸调pH至5.4),梯度洗脱,该方法于2019年修订并沿用至今,缓冲盐的浓度较高,长时间运行有可能引起盐析出,堵塞色谱柱或管路;梯度洗脱需要平衡色谱柱,用时相对较长。而在修订之前《美国药典》[8]的方法是以乙腈-冰醋酸-四丁基氢氧化铵-甲醇的混合溶液为流动相,溶液配制复杂,四丁基氢氧化铵为离子对试剂,长期运行对色谱柱和液相系统的损伤较大,且使用后会使柱平衡时间延长,平衡时间不够可能对实验的重复性造成影响;并且采用C8分析柱,相比于HPLC法最常用的C18柱,不利于方法的推广应用。文献报道[7]的HPLC法是采用内标法,需配制胺碘酮内标溶液,样品前处理溶液配制操作和结果计算相对较为耗时,流动相采用67 mmol·L-1磷酸盐缓冲液,浓度较高,对色谱系统的长期运行有一定损害。本研究采用乙腈-20 mmol·L-1磷酸二氢铵溶液为流动相,缓冲盐浓度较低,有利于液相系统的长时间运行和样品的连续测定,与用更高浓度的缓冲盐进行比较,结果显示20 mmol·L-1磷酸二氢铵溶液即可获得较好的峰形。采用等度洗脱的方式能够节省时间,可在10 min之内完成测定,经专属性试验,样品中的辅料和强制降解产物不干扰目标成分的定量分析。采用外标法定量,相比于内标法,样品前处理溶液配制过程以及测定结果计算简便快捷。
猜你喜欢
云南化工(2023年8期)2023-08-21
今日消防(2023年6期)2023-07-27
煤化工(2022年3期)2022-07-08
云南化工(2021年11期)2022-01-12
广州化工(2020年8期)2020-05-19
安徽农业科学(2019年13期)2019-08-27
中国资源综合利用(2016年10期)2016-01-22
华东理工大学学报(自然科学版)(2015年5期)2015-02-27
河南科技(2014年23期)2014-02-27
化学与生物工程(2011年4期)2011-07-25
