
鲍鱼脏器碱提多糖的分离纯化、结构表征及抗氧化活性研究
2024-02-23 07:19林志超潘晓明吴启赐黄家福潘裕添
食品工业科技 2024年4期
林志超,潘晓明,吴启赐,薛 钰,黄家福,潘裕添,*
(1.闽南师范大学菌物产业工程技术中心,福建漳州 363000;2.萌得尔(厦门)生物科技有限公司,福建厦门 361000)
鲍鱼是一种海洋单壳软体贝类,具有重要的营养价值[1],富含蛋白质[2]、脂肪[3]、糖类[4-5]、氨基酸、维生素、微量元素等营养成分,有重要的生物功能和很高的生物活性[6-7],在生命科学和医药上具有巨大的研究和应用价值[8]。而在鲍鱼的深加工过程中,占鲍鱼体重的20%~30%的脏器通常被废弃[9-11],造成经济损失和环境污染。研究表明,鲍鱼脏器多糖有广泛的生物学活性,如抗氧化[12]、抗肿瘤[13]、免疫调节、降血糖、降血脂[14]、抗病毒等[15-16]。
鲍鱼脏器多糖提取方法有水提、碱提和酶法提取等[17]。这些方法各有优缺点:水提法成本低,对多糖结构影响小,但提取率低且耗时;酶法提取效率高,条件温和,但成本高,对多糖结构影响较大;碱提法成本低,效率高,但要注意控制提取条件,pH 应迅速调中性,以防止多糖降解[18]。碱提法适用于酸性多糖的提取[19],而海洋生物多糖大都为酸性多糖[20]。但鲍鱼脏器碱提多糖鲜有报道,因此,对比鲍鱼脏器碱提多糖与其他方法提取的鲍鱼脏器多糖在单糖组成、分子量大小等结构及组成上的异同,具有一定的研究价值和意义。
本文采用热碱水浸提法和层析法制备高纯度Aavp,并利用红外光谱法、高效液相色谱、气相色谱和化学方法等分析纯化Aavp 的初级结构,最后检测其体外抗氧化活性。本文着眼于海洋生物资源的高效开发,旨在提高鲍鱼的经济附加值,充分利用废弃的鲍鱼脏器资源,为鲍鱼产业的可持续发展提供科学依据。
1 材料与方法
1.1 材料与仪器
杂色鲍(Haliotis diversicolor)福建省东山县长盛食品有限公司;单糖标准品(D-木糖、D-半乳糖、D-葡萄糖、D-甘露糖、D-阿拉伯糖和D-鼠李糖)、葡聚糖(Dextran T-10、T-40、T-70、T-110 和T-500)优级纯,美国Sigma 公司;Tris、硫酸、苯酚、乙醇、盐酸、溴化钾、乙酸酐、高碘酸钠、盐酸羟胺、吡啶、氢氧化钡、乙二醇、抗坏血酸、邻苯三酚、DPPH 等分析纯,上海阿拉丁生化科技股份有限公司。
UV-2102PC 型紫外可见分光光度计 上海光谱仪器有限公司;Senco R1002B 旋转蒸发仪 上海申生科技有限公司;SBS-100 数控计滴自动部分收集器 上海沪西分析仪器有限公司;Pellicon XL 超滤膜包 Biomax 5 kDa 0.005 m2美国Millipore 公司;7890B 气相色谱、Agilent 1200 HPLC 系统、1260 Infinity II 示差检测器 美国Agilent 公司;ÄKTA pure 蛋白纯化系统 美国GE 公司;NICOLET iS10傅里叶红外光谱仪 德国赛默飞世尔公司;TG209-F1 热重分析仪 德国耐驰公司;UV-3200PC 紫外可见光谱仪 上海美谱达仪器有限公司。
1.2 实验方法
1.2.1 Aavp 粗多糖提取 参考戴宏杰等[21]的方法加以改进。具体方法如下:从杂色鲍中剥离鲍鱼脏器,并去除鲍鱼脏器的结缔组织及脂肪组织,洗净消化道中的食物残渣,匀浆后冻干,研磨后过30 目筛备用。取10 g 冻干粉,用4 倍质量95%乙醇60 ℃下回流6 h 进行脱脂[22],重复两次,干燥,用8 倍质量超纯水在95 ℃下提取3 h,重复三次,过滤。滤渣加入100 mL 0.5 mol/L NaOH 溶液中,95 ℃水浴搅拌3 h,离心(10000 r/min,10 min)。滤渣提取2 次,合并上清液。调节上清液至pH7,经旋转蒸发浓缩得浓缩液。将4 倍质量95%乙醇与浓缩液混合过夜,离心(12000 r/min,20 ℃,15 min),沉淀用超纯水溶解,并用旋转蒸发仪脱去残余乙醇(60 ℃)。再用超滤(5000 Da)除盐浓缩,Sevage 法去除蛋白质[23],旋蒸去除有机溶剂,冻干备用。
1.2.2 多糖含量及多糖得率计算 用苯酚-硫酸法测定多糖含量[24],其标准曲线回归方程为y=0.0125x+0.0625,R2=0.9983。分别按公式(1)和(2)计算多糖含量及多糖得率。
式中:D,纯化后冻干粉多糖含量,%;c,样品溶液多糖浓度,mg/mL;v,溶液总体积,mL;n,稀释倍数;m1,鲍鱼脏器多糖冻干粉质量,g。
式中:G,鲍鱼脏器冻干粉多糖得率,%;c,样品溶液多糖浓度,mg/mL;v,溶液总体积,mL;n,稀释倍数;m2,鲍鱼脏器冻干粉质量,g。
1.2.3 Aavp 纯化
1.2.3.1 DEAE Sepharose Fast Flow(DFF)柱层析参考林志超等[25]方法加以改进,确定层析方法如下:称取5 g 粗多糖Aavp,充分溶解于40 mL Tris-HCl(pH7.2,50 mmol/L)溶液,0.2 μm 微孔滤膜过滤,上样至DFF 阴离子交换柱(柱体积为Φ 5.0 cm×25 cm),用0.5 mol/L NaCl 溶液梯度洗脱,流速为2 mL/min,收集洗脱液,检测多糖含量。以管数为横坐标,多糖含量为纵坐标,绘制洗脱曲线。根据洗脱曲线收集主峰,减压旋转蒸发浓缩,真空冷冻干燥备用。
1.2.3.2 Sephacryl S-400 HR 柱层析 经过预实验,确定层析方法如下:采用0.2 mol/L NH4HCO3溶液在流速30 mL/h 下平衡Sephacryl S-400 HR 柱(柱体积为Φ 2.6 cm×90 cm)24 h 后,40 mg 通过上述DFF 柱层析获得的多糖组分溶解于1 mL 0.2 mol/L NH4HCO3溶液后上样,收集洗脱液并检测多糖含量,绘制洗脱曲线,收集洗脱曲线主峰,减压旋转蒸发,超纯水透析(4 ℃),至超纯水中检测不出Cl-(AgNO3沉淀法[26]),真空冷冻干燥备用。
1.2.4 Aavp 初级结构分析
1.2.4.1 Aavp 纯度检测及分子量测定 利用HPLC检测Aavp 组分纯度及分子量[27]。多糖标准溶液:Dextran T-10、T-40、T-70、T-110 和T-500,浓度为10 mg/mL。HPLC 色谱条件:Agilent ZORBAX GF-250 分析柱,流速0.3 mL/min,柱温65 ℃,流动相为超纯水,采用示差检测器检测,温度65 ℃,进样体积20 μL。以标准样品色谱峰的保留时间对Mw 的对数进行回归,得回归方程。将Aavp 样品的保留时间代入回归方程,得Mw。
1.2.4.2 紫外光谱分析 将多糖样品配制成0.5 mg/mL 的水溶液,在200~500 nm 波长范围内进行紫外波长扫描[28]。
1.2.4.3 红外光谱分析 将1 mg 干燥的纯化多糖样品和100 mg 溴化钾研磨均匀,压成透明薄片后,上傅里叶红外光谱仪分析,扫描波数为400~4000 cm-1[29]。
1.2.4.4 单糖组成分析 称取200 mg 纯化Aavp 样品,溶于4 mL 1 mol/L 的H2SO4溶液,封管,100 ℃水浴反应8 h 后,用饱和Ba(OH)2溶液中和,离心(3000 r/min,10 min),上清液浓缩至4 mL,再离心(12000 r/min,10 min)去除沉淀物,上清液真空冷冻干燥,备用。称取10 mg 上述冻干样品和系列单糖标准品,加入10 mg 盐酸羟胺和0.5 mL 吡啶,90 ℃水浴30 min,冷却至室温,再加0.5 mL 乙酸酐,90 ℃水浴30 min,样品经0.45 μm 有机膜过滤后进行GC分析。
气相色谱条件:A.T.农残1 号毛细管柱(Φ 0.53 mm×30 m);氢火焰离子化检测器(FID);气化室温度270 ℃;柱温恒温220 ℃;检测器温度300 ℃;气体流速:载气N2为20 mL/min;H2为30 mL/min;空气为200 mL/min;进样量1 μL。
1.2.4.5 糖苷键测定 采用张惟杰[30]的方法加以修改。
标准曲线绘制:分别吸取0、0.5、1.0、1.5、2.0、4.0 mL 浓度为15 mmol/L NaIO4标准溶液于试管中,加纯水至4 mL。从上述各管中分别吸取20 μL溶液,加水定容至5 mL,在223 nm 下测定吸光度。以NaIO4溶液浓度为横坐标,吸光度为纵坐标,绘制标准曲线。
高碘酸氧化:称取样品205 mg,加入150 mL NaIO4溶液,避光反应。于4、24、48 h 等间隔时间取样稀释250 倍后,223 nm 测定吸光度值。待其稳定后,加入过量乙二醇并搅拌30 min。吸取2 mL 样液,以酚酞为指示剂,用0.01 mol/L NaOH 溶液标定,以NaIO4溶液为空白对照测定HCOOH 产生量。剩余样液加入过量KBH4,避光反应18~24 h。将经上述处理后的样液用5000 Da 超滤膜超滤去除过量KBH4,冻干得多糖醇干品。
Smith 降解:取多糖醇10 mg 加入5 mL 2 mol/L硫酸溶液,酒精喷灯封管,100 ℃水浴8 h。结束后,加入Ba(OH)2粉末中和,过滤,滤渣用少量水洗涤,合并滤液与洗液,冻干。
气相分析:方法同1.2.4.4。
1.2.4.6 热重分析 通过热失重分析仪测定Aavp 的热稳定性。条件为:N2流速:50 mL/min;温度范围:28~900 ℃;升温速率:10 ℃/min。
1.2.5 抗氧化活性测定
1.2.5.1 多糖体外清除O2-·能力测定 根据王丽娟等[31]的方法进行适当改进。用超纯水配制不同浓度的抗坏血酸,分别为0.2、0.4、0.6、0.8、1.0 mg/mL;同时分别配制0.2、0.8、1.6、3.2、6.4 mg/mL 的Aavp 粗多糖溶液和Aavp IIa 溶液。取2.55 mL Tris-HCl 溶液(pH8.2,0.05 mol/L),25 ℃恒温预热20 min,加入0.4 mL 上述不同浓度的抗坏血酸溶液、Aavp 粗多糖溶液和Aavp IIa 溶液,再加入10 mmol/L 邻苯三酚0.05 mL,振荡摇匀,反应4 min后立即加入50 μL 10 mol/L HCl 终止反应,325 nm处测定其吸光度。按公式(3)计算清除率。
式中:B,O2-·清除率,%;A0,空白组在波长325 nm 处的吸光值;Ai,试样组在波长325 nm 处的吸光值。
1.2.5.2 多糖体外清除DPPH·能力测定 抗坏血酸和Aavp IIa 分别用1.2.5.1 方法配制成溶液。将2 mL 0.2 mmol/L DPPH·溶液(无水乙醇溶解)加入2 mL 不同浓度上述的抗坏血酸和Aavp IIa 溶液中,混匀,室温避光反应30 min,在517 nm 下测定吸光度。按公式(4)计算DPPH·清除率。
式中:C,DPPH·清除率,%;A0,空白组在波长517 nm 处的吸光值;Ai,试样组在波长517 nm 处的吸光值。
1.3 数据处理
试验重复三次,利用Design Expert 8.0.6 进行数据统计分析,采用Origin 2017 软件绘图,SPSS 26.0软件进行显著性分析(P<0.05)。
2 结果与分析
2.1 Aavp 的分离纯化
2.1.1 DEAE Sepharose Fast Flow 纯化 经过方法
1.2.3.1 处理,Aavp 得率为11.21%。Aavp 经DFF 层析柱分离后,分别得Aavp I 和Aavp II(图1),得率分别为3.08%和61.63%。经真空冷冻干燥后,获得Aavp I 和Aavp II 为质地较硬的白色晶体,可能是洗脱液中盐分未除去,进一步采用尺寸排阻层析分离。
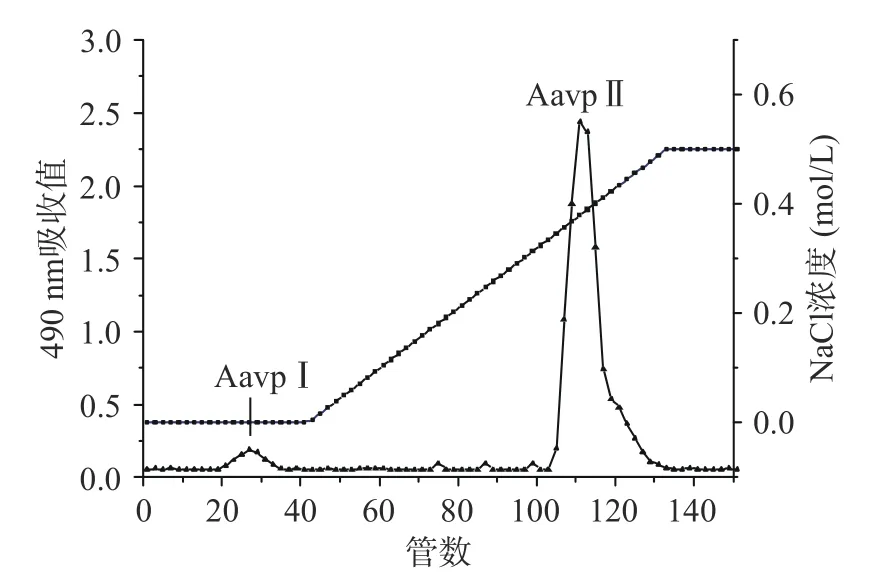
图1 Aavp 的DEAE Sepharose Fast Flow 柱层析洗脱曲线Fig.1 Elution curve of Aavp on a DEAE Sepharose Fast Flow column
2.1.2 Sephacryl S-400 HR 纯 化 Aavp I 和Aavp II 分别进行Sephacryl S-400 HR 层析,结果如图2所示。Aavp Ia 得率及多糖含量分别为78.62%和90.99%,呈白色絮状固体,可能是因为小分子盐大部分被洗脱,剩下主要是大分子多糖,不易结晶,从而呈絮状。Aavp IIa 得率及多糖含量分别为90.34%和98.66%,也呈白色絮状固体。Aavp Ia 保留时间和Aavp IIa 相差不大,说明两者分子量可能较为接近。结合图1 和图2 可看出,Aavp II 组分得率较高,而组分Aavp IIa 得率也高于组分Aavp IIb。因此,选择Aavp IIa 进行结构初步分析。
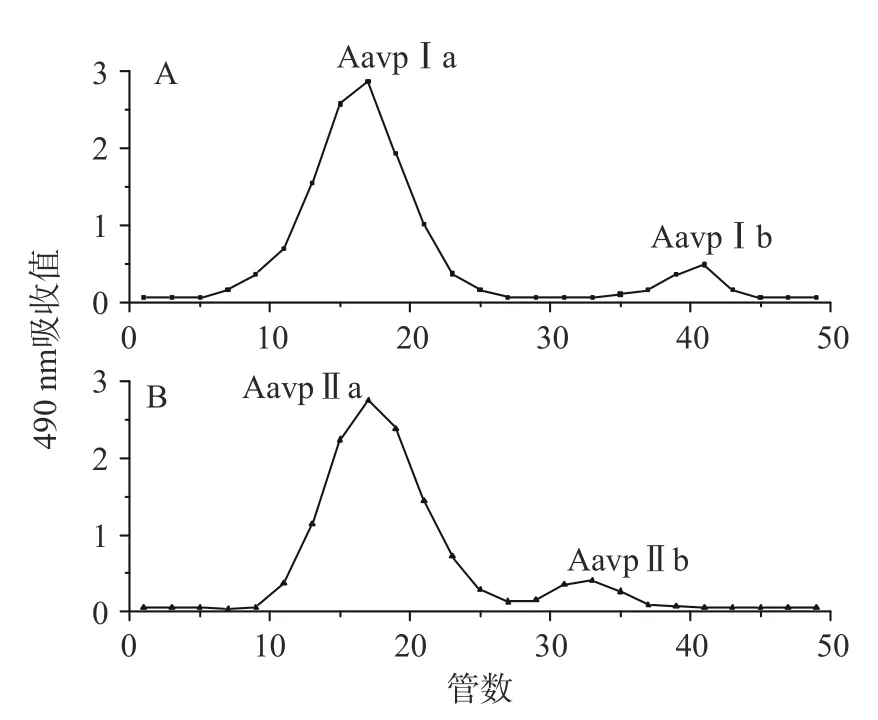
图2 Aavp 的Sephacryl S-400 HR 柱洗脱曲线图Fig.2 Elution curve of Aavp on a Sephacryl S-400 HR column
2.1.3 Aavp IIa 纯度和分子量 由图3A 可得回归方程:lgMw=32.254-4.4177tR,R2=0.9902。如图3B所示,Aavp IIa 在9.1872 min 时具有一个对称尖锐单峰,说明该组分纯度高。将Aavp IIa 保留时间代入方程,得Aavp IIa Mw 为166513 Da。与程婷婷等[32]的结果相比,其鲍鱼脏器多糖分子量约为3×105Da,显著高于Aavp IIa,这可能是因为程婷婷采用的酶解法与本文的提取方法不同。
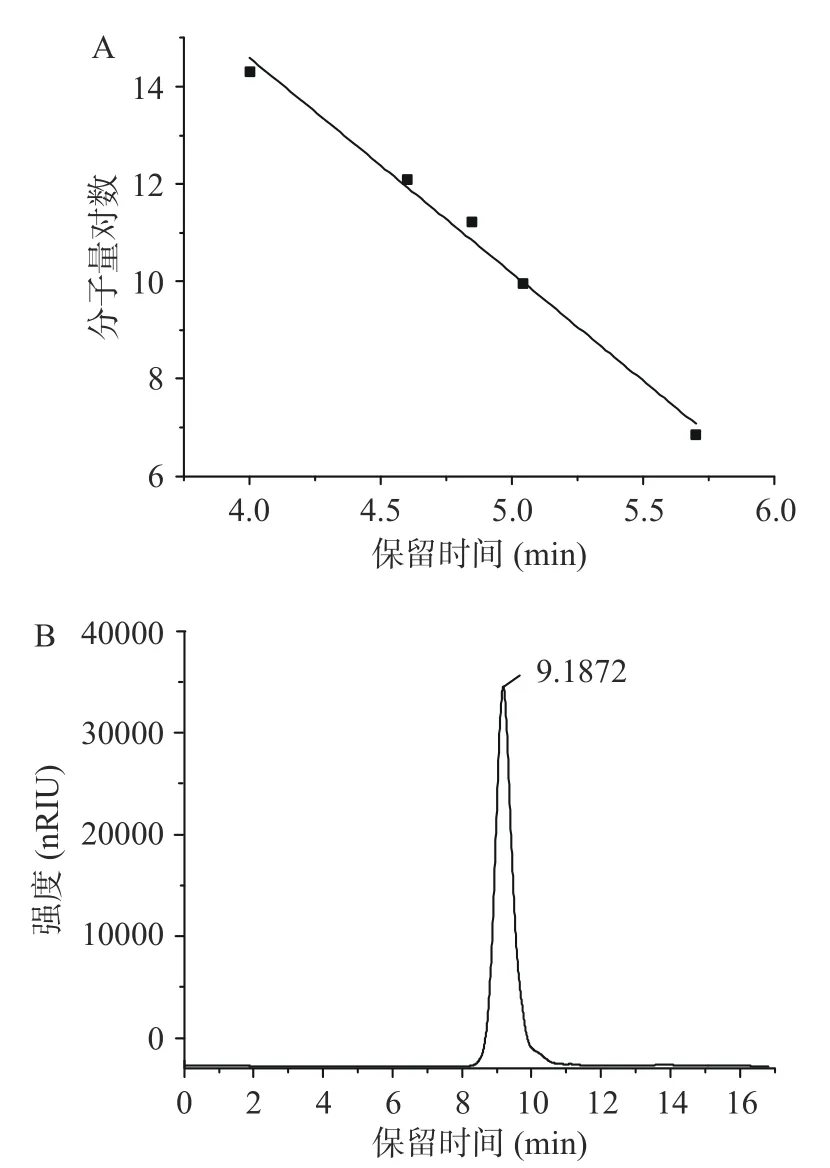
图3 分子量标准曲线(A)和Aavp IIa HPLC 图谱(B)Fig.3 Standard curve of molecular weight (A)and HPLC spectrum of Aavp IIa (B)
2.2 Aavp IIa 的初级结构分析
2.2.1 紫外光谱分析 由图4 可知,Aavp IIa 在260和280 nm 处没有紫外吸收,而粗多糖有紫外吸收,说明经过纯化后的Aavp IIa 基本不含核酸和蛋白质。
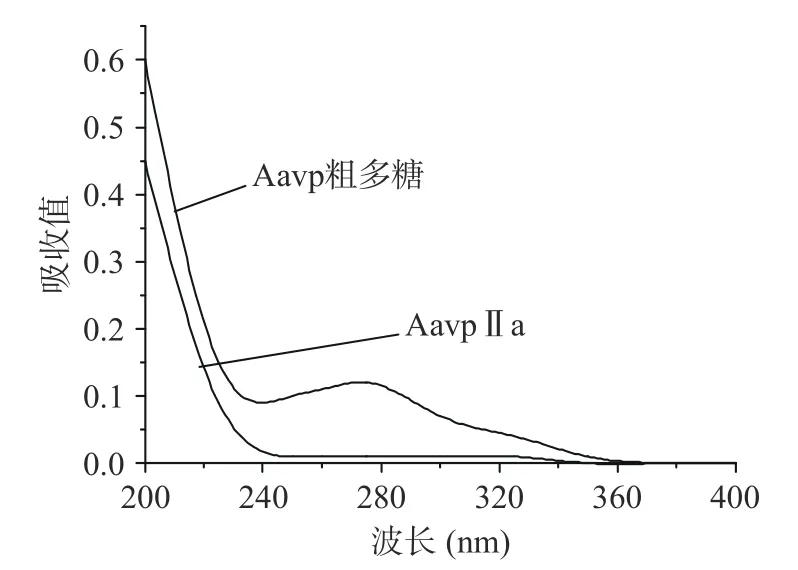
图4 鲍鱼脏器碱性多糖紫外图谱Fig.4 Ultraviolet spectra of Aavp
2.2.2 红外光谱分析 Aavp IIa 与溴化钾混匀压片后进行红外光谱分析,如图5 所示,Aavp IIa 具有典型的多糖光谱信号[33],3367.1 cm-1峰是O-H 伸缩振动造成的;2892.7 cm-1峰是C-H 伸缩振动造成的;1643.05 cm-1峰是-CHO 的C=O 伸缩振动造成;Aavp IIa 在1730~1700 cm-1不产生吸收峰,表明其不含糖醛酸或者是含量极少[34];1369.21 与1419.35 cm-1是 C-H 变角振动造成的;1029.73、1076.08 和1155.115 cm-1处的吸收峰说明有吡喃糖单元结构片段[28];858.17 cm-1处存在吸收峰,说明Aavp IIa含有α-型吡喃多糖[35]。本红外图谱与李冬梅等[36]的红外图谱相比,都具有典型多糖信号,但是没有硫酸基吸收峰,说明两种多糖还是存在一定的差别。
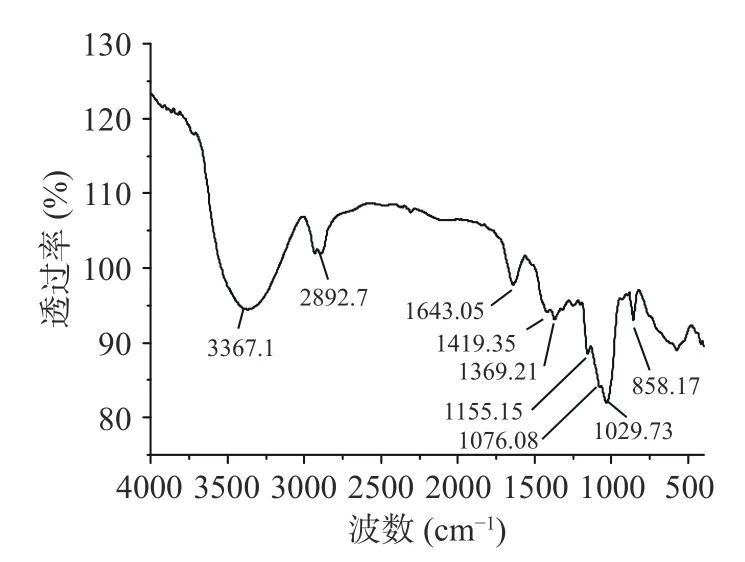
图5 Aavp IIa 傅里叶红外图谱Fig.5 Infrared spectra of Aavp IIa
2.2.3 单糖组成分析 单糖标样的保留时间如下:木糖(xyl)1.529 min、葡萄糖(glu)2.253 min、半乳糖(gal)2.361 min 和阿拉伯糖(ara)1.506 min。由表1可知,组分Aavp IIa 有两个峰,保留时间分别为1.53 和2.359 min,可推知Aavp IIa 单糖组分分别为木糖和半乳糖。根据峰面积推算,两者摩尔百分比分别为43.91%和56.09%。李冬梅等[36]的研究结果显示,皱纹盘鲍脏器多糖的单糖组成为岩藻糖、半乳糖、葡萄糖、木糖和鼠李糖,与本文有所差异。这可能是因为李冬梅是用碱性蛋白酶解法获得皱纹盘鲍脏器多糖,而本文则用碱处理水煮的滤渣制备得碱提多糖,从而造成Aavp IIa 的单糖组分较少的情况。

表1 多糖组分Aavp IIa 单糖组分分析Table 1 Analysis of monosaccharide component of Aavp IIa
2.2.4 糖苷键测定结果 Aavp IIa 经高碘酸氧化处理,每摩尔己糖残基消耗2.694 9 mol 高碘酸,没有甲酸产生。对图6 分析,产物中各个组分摩尔百分比为:甘油20.20%,赤藓醇11.81%,木糖33.85%,半乳糖34.14%。根据高碘酸氧化-Smith 降解原理,半乳糖残基1→4 糖苷键生成赤藓醇,1→3 糖苷键生成半乳糖,半乳糖1→2、1→6 糖苷键生成甘油;木糖残基1→2、1→4 糖苷键生成甘油,1→3 糖苷键生成木糖。根据上述数据可计算出,Aavp IIa 中半乳糖1→4 糖苷键摩尔百分比为11.81%,半乳糖1→3 糖苷键为34.14%,由于甲酸没有产生,所以Aavp IIa 中没有半乳糖1→6 糖苷键,同时可推知,半乳糖1→2 糖苷键为10.14%。Aavp IIa 中木糖1→3 糖苷键摩尔百分比为33.85%,由于木糖1→2、1→4 糖苷键的最终产物都为甘油,通过高碘酸氧化和Smith 降解无法分辨出两种糖苷键的比例,只能得到以下结论:可能性1,当只含木糖1→4 和木糖1→3 时,木糖1→4 摩尔百分比为10.06%;可能性2,当只含木糖1→2 和木糖1→3 时,木糖1→2 摩尔百分比为10.06%;可能性3,当含有木糖1→4、1→2 和木糖1→3 时,木糖1→4 和木糖1→2 总的摩尔百分比为10.06%。

图6 单糖标样及Aavp IIa 的Smith 降解产物GC 图谱Fig.6 Gas chromatogram spectrum of monosaccharide standard samples and Smith degradation products of Aavp IIa
2.2.5 热重分析 从图7 可知,Aavp IIa 的热失重有三个阶段:第一阶段是在室温到102.4 ℃之间,这一阶段的热失重主要是水分的丢失引起的,热失重率约为11.59%;第二阶段为Aavp IIa 的急速热分解,在302.4 ℃热失重达到最大,拐点在226.4 ℃,这阶段的热失重率为43.65%,说明此时多糖分子的化学键被破坏,多糖正在被分解;第三阶段为Aavp IIa 的缓慢热分解,拐点在332.6 ℃,这阶段的热失重率为30.89%。热重结果显示Aavp IIa 多糖分子在226.4~332.6 ℃剧烈分解,说明Aavp IIa 可在226 ℃以下保持较好的稳定性。

图7 Aavp IIa 热重图Fig.7 TGA curves of Aavp IIa
2.3 Aavp IIa 的抗氧化活性分析
2.3.1 体外清除O2-·能力 如图8 所示,Aavp IIa 有较强的清除能力,但要弱于抗坏血酸(VC)。当浓度从0~6.5 mg/mL 时,Aavp IIa 的浓度与其清除能力成正相关。当Aavp IIa 的浓度为6.5 mg/mL 时,其对O2-·清除能力最强,达到85.89%。相对地,Aavp粗多糖的消除能力要比Aavp IIa 低,只有30.51%。罗晓航等[37]酶法提取的鲍鱼脏器粗多糖浓度低于10 mg/mL 时,几乎无自由基清除能力;浓度为20 mg/mL 时,清除率也仅为25.4%。这说明碱提多糖对于O2-·清除能力要好于酶法提取的鲍鱼脏器多糖。Aavp IIa 多糖分子具有还原性的半缩醛羟基,当其接触到O2-·之后,就会发生氧化还原反应[38]。

图8 Aavp IIa 和Aavp 对O2-·的清除能力Fig.8 Scavenging ability of Aavp IIa and Aavp on superoxide anion free radical
2.3.2 体外清除DPPH·能力 多糖作为大分子化合物可以为DPPH·提供质子,从而生成稳定的化合物。因此,多糖具有清除DPPH·的可能,可在517 nm波长处出现吸收减弱,吸收程度与清除活性有关[39]。如图9 所示,当浓度从0~6.5 mg/mL 时,Aavp IIa 的浓度与其对DPPH·清除能力成正相关。当Aavp IIa 的浓度为6.5 mg/mL 时,其对DPPH 清除能力最高,为62.17%。这说明Aavp IIa 有一定的DPPH 自由基清除能力,但要弱于抗坏血酸(VC),而Aavp 粗多糖的清除率为25.39%。陈胜军等[40]提取的10 mg/mL 鲍鱼内脏多糖的DPPH·的清除率可达97.39%,其清除能力要高于Aavp IIa。这可能是由于Avap IIa 提供电子的能力弱于陈胜军提取的鲍鱼脏器多糖[41]。

图9 Aavp IIa 和Aavp 对DPPH·的清除能力Fig.9 DPPH· scavenging ability of Aavp IIa and Aavp
3 结论
本研究采用热碱水浸提法和柱层析法分离制备Aavp,并利用HPLC、GC、红外光谱、热重分析及高碘酸氧化-Smith 降解等方法对其进行结构表征。结果显示,粗多糖经DEAE Sepharose Fast Flow 和Sephacryl S-400 HR 层析,可得Aavp Ia、Aavp Ib、Aavp IIa 和Aavp IIb 等4 种组分,其中Aavp IIa 得率最高,为90.34%,多糖含量为98.66%。对得率最高的Aavp IIa 组分进行表征,分子量为166513 Da,由木糖和半乳糖组成,其摩尔比43.91 : 56.09,为α-型吡喃多糖,在226.4~332.6 ℃范围内剧烈分解,热失重率为43.65%。糖苷键类型可能为:a.半乳糖1→4 糖苷键摩尔百分比为11.81%,半乳糖1→3 糖苷键为34.14%,半乳糖1→6 糖苷键为0,半乳糖1→2 糖苷键为10.14%;b.木糖1→3 糖苷键摩尔百分比为33.85%,木糖1→4 和木糖1→2 总的摩尔百分比为10.06%。Aavp IIa 对O2-·的清除率为85.89%,对DPPH·的清除率为62.17%,体现了良好的抗氧化能力。本研究采用热碱提取法制备鲍鱼脏器碱提多糖,并分析其抗氧化活性,为开发利用鲍鱼脏器等副产品提供理论依据,也为鲍鱼脏器碱提多糖的生物活性研究提供初步证据,后续研究将聚焦于鲍鱼脏器碱提多糖其他的生物学活性进行研究。
猜你喜欢
现代食品科技(2023年12期)2024-01-09
意林彩版(2022年2期)2022-05-03
意林彩版(2022年4期)2022-05-03
文萃报·周二版(2019年2期)2019-09-10
天然产物研究与开发(2019年1期)2019-03-01
作文·初中版(2017年8期)2017-09-04
天然产物研究与开发(2016年1期)2016-06-05
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
分析科学学报(2015年3期)2015-10-18
四川师范大学学报(自然科学版)(2014年4期)2014-10-09
