
阳离子对 8-氧 -7,8-二氢 -2′-去氧鸟嘌呤核苷构型的影响
2014-10-09 01:19:56梁晓琴李来才
四川师范大学学报(自然科学版) 2014年4期
郑 妍, 梁晓琴, 李来才
(四川师范大学化学与材料科学学院,四川成都610066)
1 研究背景
去氧鸟苷是DNA的基本组成之一,由一个鸟嘌呤碱基和一个去氧核糖通过N—糖苷键连接而成.活性氧(ROS)进攻鸟嘌呤导致几种氧化产物,其中最容易进攻的位置是C8原子,而相应的氧化产物是8-氧-7,8-二氢鸟嘌呤(8-oxoG).在DNA复制时,8-oxoG可直接引起碱基对由G∶C到T∶A转化,因为8-oxoG具有与腺嘌呤配对的趋势[1].8-oxoG被普遍认为是引起变异发生、致癌作用和老化的高度危险因素之一,因此,8-oxoG在过去的数十年里受到了广泛的关注[2-3].在8-氧-7,8-二氢-2′-去氧鸟嘌呤核苷(8-oxodG)中,N—糖苷键连接着8-oxoG碱基和去氧核糖糖环.N—糖苷键的断裂和水解是其碱基剪切修复(BER)中最重要的过程,也由此维持储存在基因中的遗传信息的完整性.人体8-羟基鸟嘌呤DNA糖苷酶(hOGG1)能选择性地识别8-oxoG∶C碱基对,并破坏8-氧-2′-去氧鸟苷(8-oxodG)中8-oxoG碱基与去氧核糖环间的N—糖苷键从而将氧化形式的8-oxoG敲除[4-5].这样的反应依赖于特殊糖苷键的活性,具有重要意义的是研究8-oxodG中N—糖苷键的内在性质和评估某些因素,比如氧化、质子化和阳离子化等对N—糖苷键稳定性的影响.近年来,有许多实验对核苷中N—糖苷键性质进行了研究.比如,N—糖苷键的非酶催化水解反应,环境和化学物质(如:H+,金属离子,烷基化的化合物)对N—糖苷键的影响.这些研究旨在了解N—糖苷键水解的内在化学性质.最近,针对这方面进行的理论计算有所增加.M.Hotokka等[6]用半经验和从头算法HF/3-21G方法计算了腺苷及其衍生物的电子分布、质子亲和力和N—糖苷键的异裂,R.Rios-Font等[7]用量子化学的 B3LYP、MP2和CCSD(T)方法研究了氧化、质子化和用Cu+和Cu2+阳离子化对2′-去氧鸟苷中N—糖苷键强度的影响;他们的研究结果表明,鸟嘌呤上引入正电荷将抑制N—糖苷键的均裂过程,但是在很大程度上支持N—糖苷键的异裂过程.有关8-oxodG的阳离子化的理论研究仍不足.选取Na+是因为这个离子非常普通并且是在生物体内与生物学最相关的金属阳离子,存在于富含AT序列的小空穴中[8].本文重点研究8-oxodG的阳离子化(H+和Na+)对N—糖苷键的构型和稳定性的影响.
2 计算方法
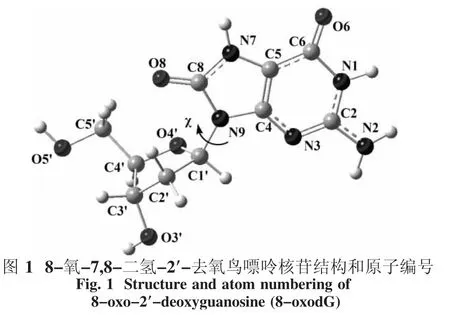
依照以前报道的类似2′-去氧鸟苷的结构[9],计算模型和原子编号见图1.在这个模型中,碱基相对于糖环来说处于反式(90.0°≦χ≦270.0°,表示二面角O4′-C1′-N9-C4的值).事实上,在8-oxodG的碱基环上,有3个位点,即N3、O6和O8原子,可与质子和钠离子结合,由此将在这3个位点引起竞争.本文中研究的化合物包括碱基环上所有可能的单质子化和阳离子化衍生物.气态中,用密度泛函方法和6-311++G(d,p)基组对化合物进行结构优化[10].M05方法用于开壳层体系的计算,稳定点的性质通过在相同水平上的振动频率分析来确认:每个稳定点的所有频率都为正.在298.15 K和一个标准大气压下通过频率分析也得到零点能(ZPVE).在相同水平下,应用自然键轨道(NBO)[11]计算了自然原子电荷,并用极化连续模型[12-13]计算了溶剂效应,用气态优化结构考虑水作为溶剂.所有的计算用Gaussian09程序包计算[14].
键的断裂通常遵循以下2种典型的过程,即均裂和异裂过程:
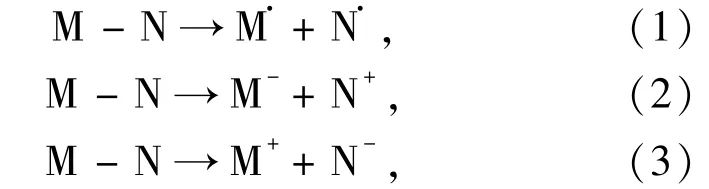
这里,(1)式表示均裂过程,而(2)和(3)式表示异裂过程.就8-oxodG及其衍生物而言,M和N分别代表8-oxoA或相应的衍生物和去氧糖环.然而,N—糖苷键以(3)式的方式异裂在能量上并不占优势,因此在本文中没有考虑(3)式的异裂.
3 结果与讨论
本研究工作以8-氧-7,8-二氢-2′-去氧鸟嘌呤核苷为研究对象,考虑了质子化和阳离子对8-oxodG的构型和N—糖苷键稳定性的影响.研究结果表明,质子化和阳离子降低了N—糖苷键的稳定性,有利于N—糖苷键的断裂,且支持异裂过程.所研究体系的优化结构及部分结构参数如图2所示,而相关的部分重要结构参数列于表1和2列出了8-oxodG的质子化产物离解能.为叙述方便,文中采用了下述符号:a代表8-oxodG的质子化产物,而b则代表8-oxodG与阳离子的络合物,而碱基上3个配位点N3、O6和O8原子分别用3、6和8表示.例如,b3表示Na+通过与N3原子相互作用得到的8-oxodG-Na+的络合物.
3.1 各物质的几何结构分析如图2~3所示,8-oxodG与其衍生物在结构上表现出显著的差别,以下的讨论都是以8-oxodG这个物质为参照物进行的.在a3和b3中,Na+(或H+)同时与N3和O4′原子相互作用,Na—N3和Na—O4′键长分别约为0.225 2和0.226 9 nm;而H—N3和H—O4′键长分别约为0.102 6和0.191 7 nm.N3和O4′原子的孤对电子分别与Na+(或H+)的空轨道相互作用,分别导致 N3—N2、N3—C4、C1′—O4′和 C4′—O4′等键减弱,键长增长;结果,在b3中,C1′原子和N9原子的相互作用增加,使N9—C1′键强度增强,键长缩短(△d=-0.001 1 nm).同时O4′原子和钠离子的相互作用也引起去氧核苷构型的改变和糖环构型的改变.分子内相互作用,N3—H(Na)…O4′,导致N—糖苷键的扭转而使化合物由初始物的反式结构转变为最终的顺式结构,其χ值在a3和b3中分别为328.3°和 281.3°.

表1 N—糖苷键的分析结果Table 1 Structural parameters of N—glycosidic

在a8和b8中,Na+(或H+)分别与O8′和O4′相互作用.当质子以顺式或钠离子与O8原子结合,存在分子内的相互作用,即 O8—H(Na)…O4′,同时O4′原子也会转移出部分电子密度.因此,O4′原子和C1′原子间的相互作用减弱,这也导致C1′—O4′键键长增加(b8)或几乎保持不变(a8).另一方面,分子内的相互作用也引起N—糖苷键的改变.例如,在a8中,N9—C1′键增长0.003 0 nm,而在b8中,N9—C1′键缩短了0.001 2 nm.这些变化与2个因素有关:体系引入正电荷和O4′原子与正离子间的相互作用.
如图2和3所示,除了以上讨论的差别外,核苷衍生物与核苷相比较仍有一些规律可循.由于引入正电荷,N9—C1′键键长(表2)增长,其变化范围从0.001 2 nm(b6)到0.002 8 nm(a8′)(除 b8 和b3外).对糖环而言,首先,C4′—O4′键键长增长而C1′—O4′键键长缩短(除 b8 和 b3 外).其次,碱基相对于糖环来说是反式,相应的χ值在187.5°~258.5°之间.
8-oxodG衍生物相对于8-oxodG的变化主要归结于以下几个因素:一方面,在去氧糖环中,“异头碳影响”和反平面孤对假设(ALPH)有利于电子流从糖环向离去基团偏移,从而促进N—糖苷键断裂.结果,N—糖苷键键长增长而C1′—O4′键键长缩短.另一方面,当碱基与质子或阳离子结合时,碱基环处于缺电子状态而成为电子受体.因此,那些与碱基环结合的富电子基团倾向于向碱基环提供电子以补偿电子不足.事实上,除了上述主要原因外,仍有其它的因素影响所研究的体系,比如,分子间相互作用、静电影响和极化影响等,并且上述变化是这些因素同时作用的结果.

表2 均裂过程和异裂过程的离解能Table 2 Dissociation energies of homolytic and heterolytic processes kJ/mol
3.2 离解能分析N—糖苷键的强度能够通过计算键的离解能评估.键离解能定义为各部分离解产物的能量和与反应物的差值.表2列出了所有研究体系的离解能.
气态时,核苷的阳离子化可以改变N—糖苷键的离解能.对8-oxodG而言,N—糖苷键均裂过程的离解能比其异裂过程的离解能低大约298 kJ/mol,因而均裂过程在气态中是首选离解方式.有趣的是,其衍生物体系的N—糖苷键异裂过程的离解能比8-oxodG的异裂过程的离解能减弱293~416 kJ/mol,而均裂过程的裂解能与中性物质相比则变化要小得多( |△E |≤94 kJ/mol),即对 8-oxodG的衍生物体系而言,异裂过程的离解能较低,是优势离解方式.因此,碱基团引入正离子非常有利于N—糖苷键的异裂过程从而使异裂成为首选过程而均裂过程相对较难进行.这些结果表明碱基基团的阳离子化对促进核苷中N—糖苷键的断裂起着重要的作用.
由于真实的裂解反应都是在溶剂中进行的,所以对于所有体系也考虑了溶剂化效应.溶剂化效应对离解能带来显著的变化.其中受溶剂效应影响最大的是8-oxodG.其异裂过程的离解能降低了大约440 kJ/mol,而其均裂的离解能仅仅下降了约107 kJ/mol.结果,考虑到溶剂效应,对8-oxodG而言首选裂解过程是异裂过程.而对其衍生物体系来说,虽然均裂和异裂过程的离解能也都有所下降,但是异裂过程的离解能仍然比对应的均裂过程的离解能低得多,所以前者仍然是首选的裂解过程.因此,异裂过程占据优势地位,正电荷引入能促进N—糖苷键的断裂.
4 结语
在本研究工作中,采用了DFT理论的M05方法选用6-311++G(d,p)机组对8-氧-7,8-二氢-2′-去氧鸟嘌呤核苷的构型和糖苷键的稳定性进行了研究,考虑了质子和阳离子对8-oxodG的N—糖苷键稳定性的影响.研究结果表明,质子化和阳离子降低了N—糖苷键的稳定性,有利于N—糖苷键的断裂.与8-oxodG相比较而言,其衍生物在结构上表现出显著的差别.N9—C1′键键长增长(除b3和b8外),其变化范围从0.001 2 nm(b6)到0.002 8 nm(a8′);C4′—O4′键键长增长而C1′—O4′键键长缩短(除b8和b3外).研究表明,“异头碳影响”和由于在碱基引入正电荷而引起的缺电子影响促进N—糖苷键以异裂过程进行裂解,正电荷引入对促进N—糖苷键的断裂起着重要的作用.而且,异裂过程是糖苷键断裂的首选方式,这与文献[7]的结论吻合.
[1] Francoise D,Magnar B,Luisa L,et al.Comparative analysis of 8-oxoG∶C,8-oxoG∶A,A∶C and C∶C DNA repair in extracts from wild type or 8-oxoG DNA glycosylase deficient mammalian and bacterial cells[J].DNA Repair,2003,2(6):707-718.
[2] Elisabeth L,Kyungrim K,Frederic C,et al.Transcription activities at 8-oxoG lesions in DNA[J].DNA Repair,2004,3(11):1457-1468.
[3] Barone F,Lankas F,Spackova N,et al.Structural and dynamic effects of single 7-hydro-8-oxoguanine bases located in a frameshift target DNA sequence[J].Biophys Chem,2005,118(1):31-41.
[4]Fujimoto H,Pinak M,Nemoto T,et al.Molecular dynamics simulation of clustered DNA damage sites containing 8-oxoguanine and abasic site[J].J Comput Chem,2005,26(8):788-798.
[5] Berti P J,McCann J A B.Toward a detailed understanding of base excision repair enzymes:transition state and mechanistic analyses of N—glycoside hydrolysis and N—glycoside transfer[J].Chem Rev,2006,106(2):506-555.
[6] Hotokka M,Lö nnberg H.Hydrolysis of adenosine:a semiempirical and ab initio study[J].J Mol Struct:Theo Chem,1996,363(2):191-201.
[7] Rios-Font R,Rodrí guez-Santiago L,Bertran J,et al.Influence of N7 protonation on the mechanism of the N—glycosidic bond hydrolysis in 2′-deoxyguanosine:a theoretical study[J].J Phys Chem,2007,B111(21):6071-6077.
[8] Hamelberg D,Williams L D,Wilson W D.Influence of the dynamic positions of cations on the structure of the DNA minor groove:sequence-dependent effects[J].J Am Chem Soc,2001,123(23):7745-7755.
[9] Hocquet A,Leulliot N,Ghomi M.Ground-state properties of nucleic acid constituents studied by density functional calculations 3:role of sugar puckering and base orientation on the energetics and geometry of 2′-deoxyribonucleosides and ribonucleosides[J].J Phys Chem,2000,B104(18):4560-4568.
[10]Petersson G A,Al-Laham M A.A complete basis set model chemistry (II):open-shel1 systems and the total energies of the first-row atoms[J].J Chem Phys,1991,94(9):6081-6090.
[11] Reed A E,Curtiss L A,Weinhold F.Intermolecular interactions from a natural bond orbital,donor-acceptor viewpoint[J].Chem Rev,1988,88(6):899-926.
[12]Barone V,Cossi M.Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model [J].J Phys Chem,1998,A102(11):1995-2001.
[13]Cossi M,Rega N,Scalmani G,et al.Energies,structures,and electronic properties of molecules in solution with the C-PCM solvation model[J].J Comput Chem,2003,24(6):669-681.
[14] Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian 09 A.01[S].Pittsburgh:Gaussian Inc,2009.
猜你喜欢
现代食品科技(2023年12期)2024-01-09 01:22:38
国际呼吸杂志(2019年20期)2019-11-23 08:46:08
西安文理学院学报(自然科学版)(2016年4期)2016-12-19 08:18:59
化工生产与技术(2016年5期)2016-11-07 02:27:43
China International Studies(2016年3期)2016-07-14 03:00:06
中国运动医学杂志(2016年3期)2016-07-10 12:07:23
分析测试学报(2015年9期)2015-12-17 16:44:28
分析科学学报(2015年3期)2015-10-18 02:25:40
质谱学报(2015年5期)2015-03-01 03:18:25
科技资讯(2013年7期)2013-04-29 00:44:03
