
硫醚与2-甲基-1,5-己二烯反应的机理研究
2024-01-18 13:36:48李青益刘国魁李云志韦瑶瑶王佳祺周慧敏周广丽夏其英
原子与分子物理学报 2024年3期
李青益, 石 云, 刘国魁, 冷 霞, 李云志,韦瑶瑶, 王佳祺, 周慧敏, 周广丽, 夏其英
(临沂大学 化学化工学院,临沂 276000)
1 引 言
硫醚类化合物是一些具有生物活性的分子以及天然产物的重要官能团,也是一些有机分子结构中的重要单元,其与烯烃的碳氢键加成反应的功能化产物是药物、天然产物、功能材料的重要结构单元,该合成策略是实现功能化硫醚衍生物最直接有效的途径,满足最直接和原子经济性的需求,符合绿色化学发展理念,一直以来都受到国内外有机合成化学家的高度关注[1]. 近几年来稀土金属配合物因其化学性质稳定、配位形式多样、与杂原子配位能力强在催化共轭双烯、非共轭双烯、苯乙烯、和带有杂原子官能化的烯烃的配位均聚及与其他单体的配位共聚反应中,体现出独特的杂原子亲和性、区域选择性、非对映选择性和立体选择性[2,3]. 王等人采用了半夹层钪催化剂,首次实现了醚和硫醚官能化的1,6-庚二烯的区域选择性和高度立体规整的环聚合[4]. 侯召民等人报道在半夹层钪烷基配合物作用下,可成功实现硫醚α-C(sp3)-H键对烯烃或者二烯烃的区域选择性加成,原子利用率达到100%,该反应具有良好的区域选择性及官能团兼容性,提供了一种新的方法高效制备官能化的硫醚[5]. 然而,其微观反应机理尚不明确,这在一定程度上限制了相关催化体系的顺利进展. 因此,本论文选取甲基戊基硫醚和2-甲基-1,5-己二烯分别作为硫醚小分子及非共轭二烯烃的代表,我们对此反应过程展开了详细的理论研究,从而为深入了解该类碳氢活化反应及为相关催化体系的设计开发提供理论信息.
2 计算方法和模型
所有的计算均使用Gaussian 09程序[6],几何优化和频率计算采用杂化交换相关密度泛函B3PW91[7],优化过程中没有使用任何对称性或几何约束限制. 对于非金属C、H、N和S原子采用6-31G(d)基组,对于金属Sc原子来说采用的是Stuttgart/Dresden 有效核势(ECP)及其相应的基组[8]. 为了获取更为精确的能量,采取包含弱相互作用的M06密度泛函方法[9],同时使用更高水平的基组进行单点能计算,即对C、H、N和S等非金属原子采用6-311+G(d,p)基组. 采取CPCM模型模拟甲苯(ε=2.37)的溶剂化效应[10]. 势能面上的自由能包含在气相计算中的校正值.
3 结果与讨论
实验表明,半夹心茂基稀土金属钪烷基配合物可以催化硫醚与2-甲基-1,5-己二烯进行碳氢加成反应生成环化产物.
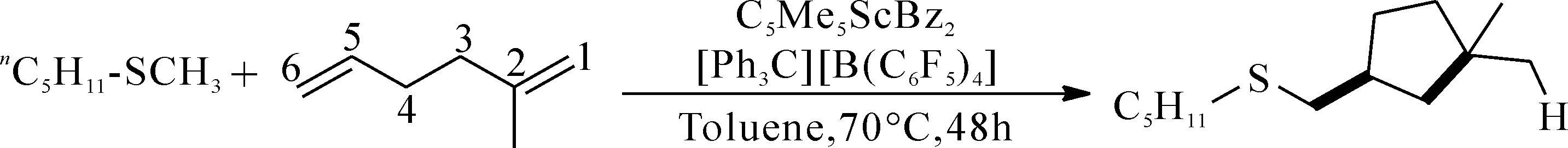
图1 钪配合物催化硫醚与2-甲基-1,5-己二烯反应Fig. 1 The reaction of thioether with 2-methyl-1,5-hexadiene catalyzed by scandium complex
3.1 活性物种的生成
如图2所示,首先甲基戊基硫醚(B)配位到阳离子型稀土金属钪配合物(A)上形成较稳定的配位络合物C,进一步由σ键交换过渡态TS[C-D]发生硫醚甲基上的C-H键活化,生成氮氮二甲基邻甲苯胺配位的三元环钪金属中间体D. 此过程需要克服的能垒为27.4 kcal/mol. 随后D释放出N,N-二甲基邻甲苯胺F,从而形成三元环钪金属中心活性物种E.

图2 钪配合物催化硫醚与2-甲基-1,5-己二烯反应活性物种生成过程Fig.2 The formation of active species via the reaction of thioether and 2-methyl-1,5-hexadiene catalyzed by scandium complex
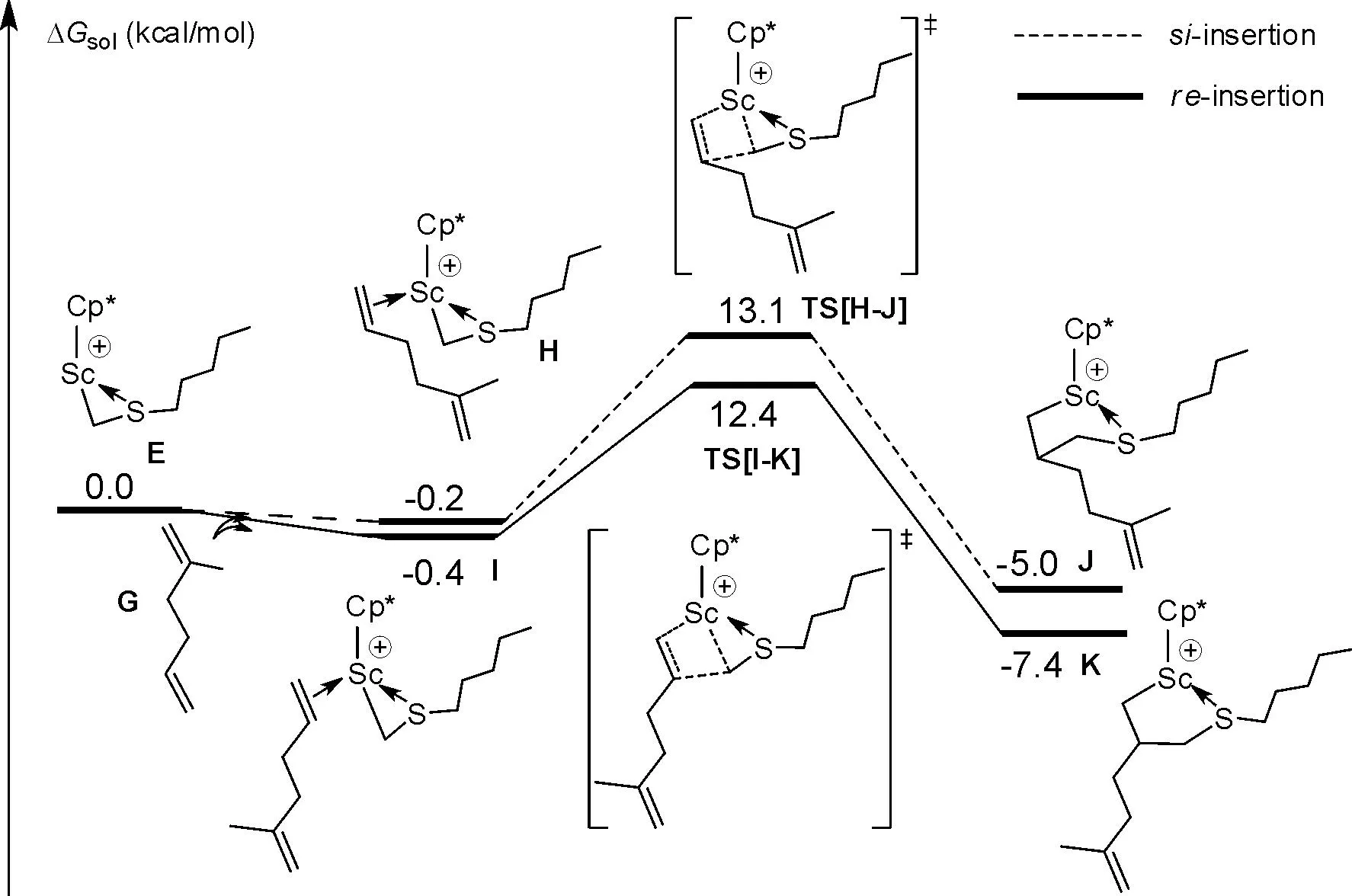
图3 2-甲基-1,5-己二烯re/si-插入过程Fig. 3 There/si- insertion process of 2-methyl-1,5-hexadiene

图4 2-甲基-1,5-己二烯分子内环化反应Fig. 4 Theintramolecular cyclization of 2-methyl-1,5-hexadiene
图5 另一分子的戊基甲基硫醚的C-H键活化过程Fig. 5 The C-H bond activation of another pentyl methyl sulfide molecule
3.2 2-甲基-1,5-己二烯的插入过程
基于生成的活性物种E分别进行模拟计算比较了2-甲基-1,5-己二烯不同插入方式所需克服的能垒大小. 首先是B以6,5-的方式配位到活性物种E,形成相应的配合络合物,经由过渡态形成稳定的金属五元环中间体,此过程需克服12.8 kcal/mol的能垒,并释放出7.4 kcal/mol能量. B以5,6-的方式配位插入需克服17.3 kcal/mol的能垒. 若B以1,2-方式进行插入,则需克服13.8 kcal/mol的能垒. 通过对比不同插入方式所形成的配位络合物、过渡态及插入中间体的能量,发现采取6,5-插入方式所形成过渡态克服能垒最低,热力学插入产物最为稳定.
3.3 2-甲基-1,5-己二烯的插入立体选择性比较
烯烃的6,5插入可能会采取re-面插入或si-面插入. 根据不同插入面的选择,进行模拟计算,结果如下图所示. 当二烯烃以si方式形成配位络合物H,随后经由过渡态TS[H-J]需要克服的能垒为13.3 kcal/mol,最后形成五元环中间体J释放能量5.0 kcal/mol.

表1 2-甲基-1,5-己二烯以不同方式插入阳离子活性物种的相对吉布斯自由能
对比发现两种插入方式在形成配位络合物时能量类似(-0.4 kcal/mol vs -0.2 kcal/mol),由配位络合物形成过渡态的过程中si-插入需要克服的能垒稍高(12.8 kcal/mol vs 13.3kcal/mol),re面插入产物比si面插入多放能2.4 kcal/mol(-7.4 kcal/mol vs -5.0 kcal/mol),更加稳定. 综上所述,6-5-re-插入是第一分子碳碳双键插入最为有利的反应路径.
3.4 2-甲基-1,5-己二烯的分子内环化反应
基于稳定的中间体K,可以进行以分子内碳碳双键配位,生成稳定的配位络合物L,放能17.2 kcal/mol,随后经历过渡态TS[L-M]形成硫醚环化中间体M,较低的环化能垒(11.9 kcal/mol)及较高的热力学稳定性(-17.4 kcal/mol),表明闭环环化过程相对比较容易,可以快速完成.
3.5 另一分子的戊基甲基硫醚的C-H键活化过程
另一分子的硫醚(B)配位到中间体M上,形成配位络合物N,并放能27.1 kcal/mol. 然后进一步通过硫醚甲基上的C-H键活化的过渡态TS[N-O],生成新的硫醚烷基化产物配位的三元环中间体O,这一过程在整个反应过程中需要克服28.6 kcal/mol的活化能垒,耗能最大,为反应的速控步. 随后释放出相应烷基化硫醚环化产物P,再生活性物种E,E将会进入下一催化循环反应.
4 结 论
本文采用密度泛函理论对硫醚与2-甲基-1,5-己二烯环化反应机理进行了详细的理论探究. 通过优化重要的中间体及过渡态,完成整个反应路径势能面,比较了反应过程活化过程和二烯烃的插入过程,探明了整个反应路径是包含活性物种的生成、非共轭二烯烃的插入、碳氢活化三部分内容的催化循环. 其次由于非共轭己二烯由于两个双键的影响,插入模式会不同,计算模拟比较分析不同插入模式(1,2-插入,5,6-插入,6,5-插入)的难易程度,以及插入过程中的立体选择性,发现其采用6-5-re-插入时需要克服的活化能垒最低,在反应时更为占优. 随后中间体进行分子内碳碳双键的插入完成关环反应. 最终是另一分子硫醚的C-H键活化,此过程需要克服的能垒最高,为该反应的速控步.
猜你喜欢
山西化工(2023年10期)2023-11-15 08:47:42
燃料化学学报(2023年3期)2023-03-11 03:34:40
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
大学化学(2021年8期)2021-09-26 10:51:16
中学课程辅导·教学研究(2021年8期)2021-07-14 13:44:52
燃料化学学报(2021年5期)2021-06-02 14:01:38
电脑知识与技术(2018年3期)2018-03-21 09:27:04
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
百科知识(2016年18期)2016-10-28 00:20:12
石油化工(2012年8期)2012-11-09 02:47:38
