
面向燃料电池双极板的不锈钢表相掺杂耐蚀性高通量计算与分析
2024-01-18 13:53张浩明徐竹田彭林法来新民
原子与分子物理学报 2024年3期
张浩明, 张 頔, 徐竹田, 彭林法,来新民
(1.上海交通大学 上海市复杂薄板结构数字化制造重点实验室,上海 200240;2.上海交通大学 机械系统与振动国家重点实验室,上海 200240)
1 引 言
质子交换膜燃料电池(Proton exchange membrane fuel cells,PEMFCs)由于其能量密度高、响应快速、环境友好等特点,被认为是具有广泛的应用前景和极高的研究价值的新一代氢能源转换装置,并且在交通运输、航空航天、能源储备等各方面取的广泛应用[1,2]. 双极板(Bipolar plate,BPP)是PEMFCs电堆中的关键部件,承担着支撑反应空间、分配反应气体和冷却水、传导电流等作用. 近些年超薄、低成本、易于大批量制造的不锈钢双极板成为燃料电池产业化的研究热点[3,4].
燃料电池极板工作于酸性(pH≈2-5)、高温(约70-80℃)、含有F-等腐蚀性离子、受电极电势作用的腐蚀性溶液环境,众多研究表明不锈钢材料受到腐蚀可能会造成接触电阻上升、离子析出(Fe、Cr离子等)毒害质子交换膜等问题[5],因此对双极板耐蚀性提出了更高要求. 目前常用的提高不锈钢材料性能的途径是采用PVD[6]、CVD[7]、PIRAC[8]、电沉积[9,10]等方法进行涂层处理,但是采用涂层改性会给双极板的生产带来额外的成本,而且涂层本身也存在开裂、脱附等失效的可能性. 因此也有学者进行了免涂层不锈钢基材的研究,通过对基材进行离子注入、成分调控等改性方式来使其满足PEMFCs性能需求,例如Feng在316L不锈钢中采用离子注入的方法注入Ni-Cr或者Ag,从而提高材料耐蚀、导电性能[11,12];Huang设计团簇式为[Ni-Fe13-xNix-1]Cr3的不锈钢体系,通过多组实验得出x约为3时具有最适合PEMFCs双极板基材的性能[13].
相较于经验指导、实验验证的传统方法,理论计算与模拟常常可以从更小的尺度上探究材料的机理并指导材料的开发,进而降低试错成本、缩短研发周期[14]. 不少学者已经开始使用理论计算方法对新型不锈钢设计展开研究,Vitos采用第一性原理方法计算了Fe100-(m+n)CrmNin合金及其加入多种金属元素后的剪切模量和体积模量,认为主元素配比为Fe58Cr18Ni24时具有适中的硬度的同时具有较好的延展性和耐蚀性,且加入少量Os或者Ir元素能进一步提高这些性能[15];Feng基于密度泛函理论(DFT)计算和加压冶金,设计了高N含量(>0.33wt%)的新型马氏体不锈钢,利用Mo合金化提高N的固溶量并减少Cr、N化合引起的Cr消耗,进而提高耐蚀性[16]. 近些年随着计算机技术的发展进步,高通量的材料计算和材料基因组的开发思路开始广泛应用于各种材料的研究中,例如具有合成或剥离可能性的2D材料[17]、高析氢反应(HER)催化性能的二元合金材料[18]、高隔热性能的ABO4白钨矿[19]、高循环寿命的锂离子电池电极材料[20]等等. 对于PEMFCs环境下的不锈钢腐蚀性质与设计改性相关的模拟计算研究,高通量方法会是高效可行的方案.
不锈钢表相在PEMFCs的酸性环境下,表相钝化层的成分以Cr2O3为主[21,22],因此本文的计算以Cr2O3为建模基础,对其进行了48种元素的掺杂替换,考虑了228种掺杂构型、102种吸附情况、453种表面空位情况,并计算了掺杂后体系的的功函数、F原子吸附能、Cr/O空位形成能等腐蚀性质,分析能够提高不锈钢钝化层在PEMFCs环境耐蚀性的元素,以期为高耐蚀不锈钢双极板基材设计提供指导.
2 计算与建模方法
2.1 计算方法
本文采用基于密度泛函理论的第一性原理计算软件VASP(Viennaabinitiosimulation package)[23]进行计算,赝势采用平面投影缀加波(Project augmented wave,PAW)[24],交换关联能采用广义梯度近似(Generalized gradient approximation,GGA)下的PBE(Perdew-Burke-Ernzerh)泛函[25],K点采样使用(4×2×1)的Monkhorst-Pack方法[26],能量收敛设置为1×10-5eV,力收敛设置为1×10-2eV,截断能设置为520 eV.
(1)替换能计算:本文中的掺杂形式主要考虑替换掺杂. 替换能是将材料中粒子替换为其他粒子的过程所需的能量,在本文中用于表征元素掺杂进入已有体系的难易程度,替换能越小表明掺杂越容易发生. 替换能的计算公式如下:
E(dis)=E(slab-X+Y)-
E(slab)-E(Y)+E(X)
(1)
式中E(dis)表示替换能,E(slab)表示未掺杂的切面模型能量,E(slab-X+Y)表示用Y元素替换切面模型中X元素后切面模型的能量,E(X)、E(Y)分别表示X、Y元素在各自稳定体系中的单原子能量.
(2)功函数计算:功函数是电子从材料内部移动到无限远处所需能量,可以表明材料内电子的稳定程度,功函数越高体系越难失去电子. 功函数的计算公式如下[27]:
φ=Φ-Efermi
(2)
式中φ表示功函数,Φ表示真空电子能量,Efermi表示材料费米能级.
(3)吸附能计算:吸附能是吸附质吸附到材料表面过程中需要的能量. 材料腐蚀模拟中计算腐蚀性粒子的在目标体系上的吸附能是研究目标体系在特定环境下耐蚀性的常用方法[28,29],吸附能的大小可以表征体系受腐蚀性粒子影响的难易程度. PEMFCs环境下破坏钝化层的腐蚀性粒子主要是F-[30,31],因此本文采用F的吸附能表征掺杂体系耐受F-腐蚀的能力. 吸附能的计算公式如下[29]:
(3)
式中E(ads)表示吸附能,E(X+slab)表示吸附X原子之后切面模型的能量,E(slab)表示吸附之前的切面模型能量,E(X2)表示孤立X2分子的能量.
(4)空位形成能计算:不锈钢PEMFCs双极板的常见腐蚀失效形式是钝化层溶解导致的点蚀[30,32],也有文献表明Cr2O3在酸性环境下且电极电位较高(约1.3V vs. SHE)时会出现溶解[21,33]. 材料溶解的难易程度常采用空位形成能来表征[34,35],空位形成能是将材料表面移除一个原子形成空位所需要的能量. 空位形成能的计算公式如下[29]:
E(vac)=E(slab-X)+E(X)-E(slab)
(4)
式中E(vac)表示空位形成能,E(slab-X)表示移除X原子后切面模型的能量,E(X)表示稳定的周期性体系中单个X原子的能量,E(slab)表示未移除原子时切面模型的能量.
2.2 建模方法

图1 (a)Cr2O3的晶胞结构;(b)Cr2O3的切面结构(左视图)Fig. 1 (a)Lattice structure of Cr2O3;(b)slab of Cr2O3 (left view)

图2 (a)Cr2O3切面对称性(俯视图);(b)选定周期的原子编号;(c)选定周期的吸附位点网格Fig. 2 (a)Symmetry of Cr2O3 slab (top view);(b)atoms’ serial number of selected period;(c)Adsorption site grids of selected period
通过对研究周期中的4个Cr原子(Cr11,Cr12,Cr21,Cr22)进行替换实现掺杂,并通过计算替换能来判断最容易发生掺杂的位点. 为了考察掺杂对于腐蚀性质的影响,选择最容易发生掺杂的位点作为掺杂位点,并且后续计算主要研究掺杂位点附近. 对于非金属元素,额外计算了对周期内O原子(O13,O14,O23,O24)替换掺杂的情况. 可以发现,Cr11、Cr22存在结构上的对称性,后续的计算也会验证这一点;同样(Cr12,Cr21)、(O11,O12)、(O21,O22)也是三组结构对称的位点.
吸附的初始位置设置在距离Cr2O3表面约2Å处,并将研究周期划分为如图2c所示网格,计算每个格点的吸附位置和吸附能,筛选出多个未掺杂时容易发生吸附的位点,对于不同的掺杂位点选取最近的易吸附位点,计算掺杂对于吸附性质的影响. 空位位点在掺杂位点周围的Cr和O中选取,分别计算其空位形成能,以其中最低值作为掺杂点附近的空位形成能. 共计有48种元素参与了掺杂计算,将有害元素或者过于活泼的元素、惰性气体元素等明显难以实现掺杂的元素排除在外.
3 计算结果与讨论
3.1 掺杂位点与类型
各元素掺杂的替换能如图3所示,横轴元素顺序为元素周期表序列顺序,替换能的大小体现掺杂发生的难易程度,替换能越低越容易发生掺杂,后续的各种耐蚀性参数的计算都基于最容易发生掺杂的位点进行. 可以发现,Cr11和Cr22位点的替换能几乎完全相同,可以验证这两个位点在结构上的对称性,同样Cr12和Cr21位点的替换能也几乎相同;对于大部分金属元素而言,都是Cr11/Cr22位点更容易发生掺杂,可能是因为Cr11/Cr22相对更接近材料表层. Mn(Cr11/Cr22:0.691 eV,Cr12/Cr21:0.649 eV)、Re(Cr11/Cr22:2.299 eV,Cr12/Cr21:2.092 eV)、Be(Cr11/Cr22:-1.339 eV,Cr12/Cr21:-1.414 eV)三种元素更容易发生Cr12/Cr21位点替换,因此这三种元素后续计算采用Cr12/Cr21位点掺杂. 另外可以看到Os、W、Tc的两种Cr掺杂位点的替换能几乎相同(两种替换能差别<1%),后续对于这些元素的计算将考虑两种掺杂位点都可能发生的情况并取平均值.
图3 Cr掺杂位点的替换能Fig. 3 Displacement energies of Cr doping sites
从过渡元素的Cr11/Cr22替换能可以发现较为明显的周期性趋势,大致以VI B族和VII B族为分界,前半部分元素Cr替换能为负,替换掺杂较易发生(Gd元素除外);后半部分元素Cr替换能为正,替换掺杂较难发生.
对非金属元素额外计算了替换O元素掺杂的替换能如图4所示,C、N、P、S、Se、Te、F元素的O替换能比Cr替换能低约2~5 eV更容易发生对O的替换,针对这部分元素的后续计算采用O位点替换掺杂;B、Si两种元素是金属与非金属之间的过渡性元素,相较于O替换更容易发生Cr的替换,后续计算采用Cr掺杂位点.

图4 非金属元素的Cr、O掺杂位点的替换能Fig. 4 Non-metal elements’ displacement energies of Cr/O
综上所述,所有参与计算的元素可以分为Cr11/Cr22、Cr12/Cr21、O21/O22、O11/O12四类掺杂:Os、W、Tc为Cr11/Cr22或Cr12/Cr21掺杂;Mn、Re、Be为Cr12/Cr21掺杂;C、N为O21/O22掺杂;P、S、Se、Te、F为O11/O12掺杂;其余元素均为Cr11/Cr22掺杂.
3.2 耐蚀性参数计算结果
(1)掺杂后功函数计算结果:
各掺杂体系的功函数计算结果如图5,未掺杂时的Cr2O3的功函数标注在图中,其中22种元素掺杂后的功函数高于掺杂前. 由于Cr2O3本身的耐蚀性已经比较优秀,可以发现无掺杂的Cr2O3的功函数相对较高,掺杂后的元素中P、S、Co、Ni、Ga、Se、Ru、Rh、Te、Ir、Pt、Au的掺杂后功函数有较明显的提升,C、N、Mg、V、Mn、Zn、Mo、Tc、Pd、In、W、OS的掺杂后元素与掺杂前变化不大,Cr2O3表相掺杂这些元素之后具有较高的稳定性.

图5 掺杂后的Cr2O3表面功函数Fig. 5 Work functions of Cr2O3 surface after doping (work function of Cr2O3 before doping is 4.7512 eV,as marked in the figure)
(2)F吸附能与掺杂后F吸附能变化量计算结果
在对研究周期划分吸附位点网格之后,计算了网格中25个位点的吸附能,其中16个位点吸附于A吸附位点(图6a中紫色位点),吸附能为-3.572 eV;6个位点吸附于B吸附位点(图6a中棕色位点),吸附能为-3.573 eV;另外3个位点吸附于其他位置,吸附能为-1.804~-2.843 eV,因此这几个位点较难发生吸附,后续只考虑A、B两个吸附点(吸附至选定研究周期外的位点平移整数个周期至选定周期内). A、B两个位点的具体吸附位置如图6b-e所示.
为研究掺杂对于吸附的影响,对于A、B两点分别选取选择与吸附位点距离最近的易掺杂位点进行替换掺杂并计算吸附能. 对于掺杂位点为Cr11/Cr22位点的元素,计算Cr11掺杂位点的A吸附位点吸附能(A点左移一个周期)和Cr22掺杂位点的B吸附位点吸附能;对于掺杂位点为Cr12/Cr21的元素,计算Cr21掺杂位点的A吸附位点吸附能和Cr12掺杂位点的B吸附位点吸附能;对于掺杂位点为O11/O12的元素,计算O12掺杂位点的A、B吸附位点吸附能(A点左移并上移一个周期);对于掺杂位点为O21/O22的元素,计算O22掺杂位点的A吸附位点吸附能(A点上移一个周期)和O21掺杂位点的B吸附位点吸附能.
掺杂后的吸附能相较于未掺杂时的变化量如图7,其中吸附能变化量为正表明相对掺杂前更难发生F的吸附,变化量为负表明F更容易吸附. 可以发现大部分元素两个吸附位点的吸附能相差不大,只有掺杂类型为O11/O12过渡金属元素掺杂的F吸附能变化量呈现出与Cr掺杂替换能、功函数相类似的周期性趋势,大致以VII B族(Mn、Tc、Re)为界,前半部分F吸附能降低,后半部分反之. 金属元素中,Co、Ni、Cu、Ge、Rh、Ag、In、Ir、Pt、Au对于F-的抑制作用较好;非金属元素中的C、N、P、S、Se、Te均对F的吸附能有所提升,但是提升幅度不大.

图 6 (a)选定研究周期内的吸附位点分布;(b)A吸附点的吸附位置(俯视图);(c)A吸附点的吸附位置(侧视图);(d)B吸附点的吸附位置(俯视图);(e)B吸附点的吸附位置(侧视图)Fig. 6 (a)Adsorption sites distribution of selected period (F atoms on purple/brown points adsorb to site A/B);(b)adsorption position of site A (top view);(c)adsorption position of site A (left view);(d)adsorption position of site B (top view);(e)adsorption position of site B (left view)
图7 掺杂后的Cr2O3表面F吸附能变化量Fig. 7 F adsorption energy change of Cr2O3 surface after doping (compared to that before doping)
(3)Cr/O空位形成能与掺杂后Cr/O空位形成能变化量计算结果:
本文分别计算了掺杂对于Cr和O的空位形成能影响,未掺杂情况下研究周期内的各位点空位形成能计算如表1,选择最低值(即最容易形成空位)作为未掺杂时的空位形成能,即Cr空位形成能为0.917 eV,O空位形成能为4.126 eV.
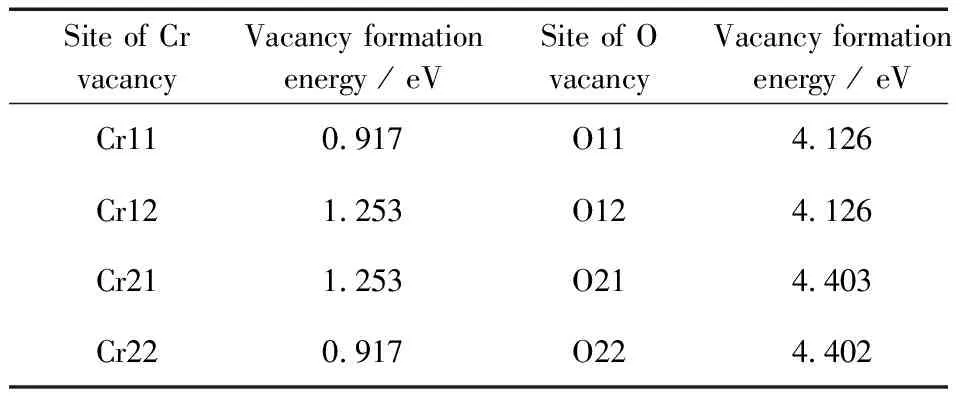
表1 未掺杂的Cr2O3空位形成能
每种掺杂位点的元素均计算其周边的Cr和O的空位形成能并取其中最低值作为空位形成能计算结果,选取范围为距离掺杂位点3.5Å以内的表层原子. 不同掺杂类型元素采用的掺杂位点和空位位点筛选范围如表2(Os、W、Tc分别考虑Cr11/Cr22和Cr12/Cr21两种掺杂类型并取其均值,不作为单独掺杂类型列出).
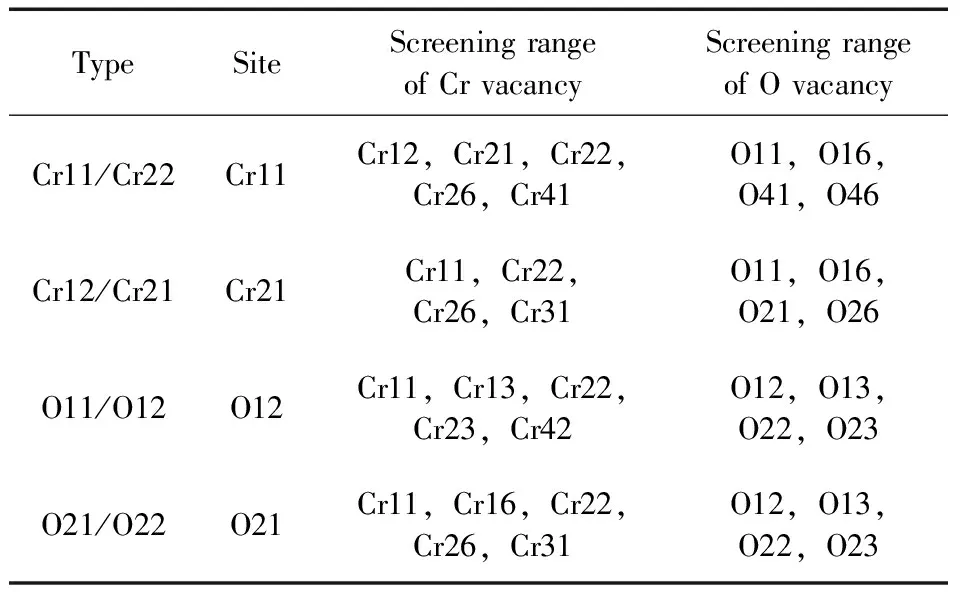
表2 各种掺杂类型选用的掺杂位点及其对应的Cr、O空位筛选范围

图8 掺杂后Cr/O空位形成能变化量及各掺杂位点对应的Cr/O空位位点Fig.8 Cr/O vacancy formation energy change after doping and corresponding doping sites of Cr/O vacancy (sites with lowest vacancy formation energy)
3.3 筛选与分析
掺杂后的F吸附能变化量、Cr空位形成能变化量、功函数的变化量的关系如图9,以抑制F吸附和Cr空位形成、高稳定性为目标,应优先考虑图中右上区域中功函数较高的元素:C、N、In、Au、Ni、Rh、Pt、Co、Ir、Ru,计算结果表明这些元素在性能上可以满足需求. 这些元素中,C、N、Ni是目前比较常见的不锈钢双极板的表面改性元素,也有不少学者对不锈钢双极板材料进行了离子注入[11,37]、渗C/N[38,39]等改性方法的探究,表明这三种元素可以提升不锈钢表面的耐蚀性. Au、Rh、Pt、Ir则是典型的贵金属元素,如果加入这些元素会导致制造成本过高,因此这四种元素的掺杂优先级较低. In、Co、Ru三种元素具有较好耐蚀性参数,且价格适中,具备掺杂的可行性,这三种元素目前较少有应用到不锈钢双极板的改性中;关于这三种元素对于不锈钢材料的作用,In2O3常被用来作为不锈钢的阴极光电化学保护材料[40],Co的合金涂层可以提高不锈钢材料的耐热酸蚀性能[41],Ru的加入则可以使不锈钢的耐蚀性能和抗应力腐蚀开裂性能得到提高[42],因此In、Co、Ru是值得尝试的不锈钢双极板改性元素;另外Co、Ru的最低Cr掺杂替换能偏高,实现掺杂的条件可能会更苛刻. 这些元素中,有文献指出Ni2+、Co2+会毒化质子交换膜中的催化剂,导致电池性能下降[5],因此,如果Ni、Co的掺杂难以将表面的耐蚀性提升至几乎不会有离子析出的程度,则应谨慎考虑选用此两种元素.

图9 掺杂后F吸附能变化量、Cr空位形成能变化量和功函数的关系Fig.9 The relationships among F adsorption energy change,Cr vacancy formation energy change and work function after doping
除了上述综合性能都较好的元素,也存在不少部分耐蚀性参数较好而某项参数不高的元素,例如Ag、Cu、Sn、Ge,除了掺杂后的功函数稍有下降外,Cr空位形成和F吸附的抑制效果均较好,其中对于Ag有学者通过离子注入Ag的方式对不锈钢双极板表面改性,使耐蚀性得到提升[12]. Mg、P、S、Zn掺杂对于F吸附能有一定提升且功函数较高,而对于Cr空位形成能的改善效果一般,其中P、S是不锈钢中典型的有害元素,且可能会对质子交换膜中的催化剂造成不利影响[5],选用优先级较低;Mg2+同样会对质子交换膜中的催化剂造成破坏,选用优先级较低.
为探究不同元素掺杂后耐蚀性变化规律的原因,对部分元素掺杂后的材料表面电子态密度(Density of states,DOS)进行了计算. 对于金属元素掺杂,耐蚀性改善效果较好的Ni和较差的Zr元素电子态密度如图10a-b,掺杂的Ni的电子主要分布于费米能级以下约2 eV至费米能级处,Ni的d电子轨道与Cr2O3中Cr、O原子的电子轨道产生杂化,表明Ni与Cr2O3基体中的原子形成了较强的键合作用[27],进而使得Cr脱离基体形成空位所需能量增加,也更难与F形成新的键合也即F的化学吸附更难以发生;对于掺杂后耐蚀性参数降低的Zr,可以发现相比于Ni的电子能态分布,Zr的d轨道电子主要分布于费米能级以上,s、p轨道电子则分别在约-50 eV和-28 eV处呈现出明显的局域化现象,表明Zr与Cr2O3中的原子未能形成较强的键合作用,进而导致了Cr空位、F吸附更容易发生,同时d轨道电子在费米能级以上的大量分布也表明相较于Ni掺杂,Zr掺杂的电子更容易逸出,这也解释了Zr掺杂后功函数的降低.

图10 掺杂后Cr2O3表面的电子态密度:(a)Ni掺杂;(b)Zr掺杂;(c)C掺杂;(d)Si掺杂Fig. 10 DOSs of Cr2O3 surface after being doped:(a)DOS of Ni-doped surface;(b)DOS of Zr-doped surface;(c)DOS of C-doped surface;(d)DOS of Si-doped surface
非金属元素中耐蚀性改善效果较好的掺杂元素C和较差的Si的电子态密度如图10c-d,与金属元素掺杂不同的是,C、Si的电子态密度分布表明这两种掺杂元素的原子轨道均与基体原子轨道发生杂化,其中C的态密度峰趋于与Cr的态密度峰重合,而Si的态密度峰趋于与O的态密度峰重合,s轨道的分布尤为明显. 这一现象主要由不同的掺杂类型导致,C采用的掺杂类型替换的是O原子,则C会趋于与原本与O成键的Cr原子成键;Si则是采用了Cr原子替换掺杂,则Si会趋于与被替换的Cr周围的O原子成键,这一成键趋势的差异导致了C和Si的引入对于Cr空位、F吸附的不同影响.
4 结 论
(2)综合比较各种腐蚀参数,C、N、In、Ru掺杂可以提高功函数、抑制F吸附和Cr空位形成,且对于质子交换膜的潜在危害较低;Au、Rh、Ir、Pt、Ni、Co掺杂的耐蚀性参数计算结果较好,但是存在价格、危害质子膜等问题. Ag、Cu、Sn、Ge掺杂对Cr空位形成和F吸附的抑制效果均较好,功函数稍有下降;Zn掺杂后的功函数数值较好且有利于抑制F吸附.
(3)从电子态密度计算结果推断掺杂原子与基体原子之间的成键作用会对掺杂后的耐蚀性产生影响,掺杂原子与Cr之间形成稳定键合是掺杂原子提升耐蚀性的重要原因.
猜你喜欢
石油管材与仪器(2020年5期)2020-11-05
陶瓷学报(2019年5期)2019-01-12
电镀与环保(2017年4期)2017-09-26
电镀与环保(2016年2期)2017-01-20
中学生数理化·高二版(2016年10期)2016-12-24
锻压装备与制造技术(2016年3期)2016-06-05
焊接(2016年9期)2016-02-27
电源技术(2015年9期)2015-06-05
读者欣赏(2014年6期)2014-07-03
语文知识(2014年2期)2014-02-28
