
激发态下甘氨酸与水分子间氢键性质研究
2024-01-18 13:36刘启帆布玛丽亚阿布力米提郑敬严
原子与分子物理学报 2024年3期
刘启帆, 布玛丽亚·阿布力米提, 向 梅, 安 桓, 郑敬严
(新疆师范大学 物理与电子工程学院, 乌鲁木齐 830054)
1 引 言
分子间氢键是位置特异的弱键,是一种重要的非共价相互作用[1,2]. 缺乏电子的氢原子与高电子密度区域之间会形成氢键,分子间氢键的形成会影响供体和受体分子的化学性质、电荷分布和结构[3,4]. 氢键在化学、物理和生物化学等各个领域都是必不可少的结构,它在生物蛋白质、DNA和RNA的结构中起着重要作用[5]. 自该理论提出后,氢键的研究得到了许多理论和实验的关注. 在实验方面,很多氢键的研究主要集中在振动(IR和Raman)光谱、疏水性、稳定性和合成过程[6-8]. 在理论研究方面,近年来发表了大量优秀的文章,主要侧重于对分子间氢键的研究以及氢键形成对分子结构的影响. 例如,Zheng通过分子动力学模拟发现木犀草素与H2O之间形成的氢键会导致木犀草素分子的电荷转移和结构变化,列举并研究了分子间氢键的九种结构[9]. Zhang在研究氢键形成对分子性质的影响时,发现聚苯胺与H2O之间的氢键会增加前者的稳定性和导电性[1].
根据电子激发态氢键动力学(ESHBD)理论[10],在激发态下,氢键的形成会导致分子的电子能谱位移,分子电荷变化等现象[11],在氢键的研究中分子间氢键激发态的性质逐渐成为当前研究的热点. Debarati Dey发现,11-苯甲酰基二苯并[a,c]吩嗪在溶剂中形成氢键后,激发态的荧光强度增加,光谱红移,性能变得更好[12]. Zhang发现激发态下分子间氢键的增强会影响聚苯胺分子从激发态到基态的内转化过程[13]. 研究激发态下分子间氢键将有助于理解分子系统的微观结构和功能,以及环境对其光物理和光化学性质的影响. 上述研究主要应用了含时密度泛函理论(TD-DFT),该理论具有较高的计算精度和效率,已被广泛用于对激发氢键性质的研究上[14,15].
氨基酸在蛋白质、RNA和DNA的形成以及人体对微量元素的吸收中起着关键作用[16]. 甘氨酸(NH2CH2COOH)作为一种简单的氨基酸,是研究激发态下分子间氢键性质的理想材料. 当水分子出现在甘氨酸分子周围时,甘氨酸的氨基(-NH2)和羟基(-COOH)将与水分子形成氢键. Muchova等对甘氨酸分子激发过程的动力学进行了理论和实验研究[17]. Bachrach模拟计算了氨基酸的稳定结构,同时对甘氨酸分子与1到7个H2O分子结合的稳定结构进行理论计算[18]. 国内也有研究团队计算了甘氨酸分子和一个H2O分子的多种结构[19,20]. 现有的研究已经证实,激发态的分子间氢键会影响分子的性质. 现有研究主要集中在对复合物甘氨酸-H2O分子氢键结构的讨论中,对氢键的描述较少,本文将分子间氢键的结构和性质作为研究重点,对比复合物激发后氢键的振动频率、强弱性质的变化,结合对氢键复合物结构的分析,了解氢键的变化规律,同时可为甘氨酸的后续研究提供理论指导.
2 理论方法和结构模型
采用DFT-b3lyp/6-311g++(d,p)水平优化甘氨酸单体分子和甘氨酸-水配合物的结构. 用上述方法计算了单体分子和复合物的电荷分布、红外光谱、前沿分子轨道(MOs)和自然键轨道(NBO)分析,以优化后的基态S0几何形状作为激发态S1计算的起点. 采用TD-DFT-b3lyp/6-311g++(d,p)水平研究了氢键络合物的激发电子结构、振动光谱、空穴-电子轨道和电子转移. 以上计算均使用Gaussian 09[21]进行.
利用AIM分析两分子间氢键的键临界点(BCP),用于研究氢键的性质. 在AIM分析中,使用Multiwfn 3.8[22]进行对BCP的计算,为了计算的准确性,对所有数据进行了DFT-D色散校正.
3 实验结果与讨论
3.1 几何结构
分子静电势可以提供分子间可能相互作用的位置信息. 甘氨酸单体的静电表面电位如图1所示. 在甘氨酸分子中,羟基中的氢原子附近有较强的正电位,羟基和氨基中的氧原子和氮原子附近有负电位. 该结果证实了羟基和氨基在甘氨酸与H2O的相互作用中发挥的重要作用.
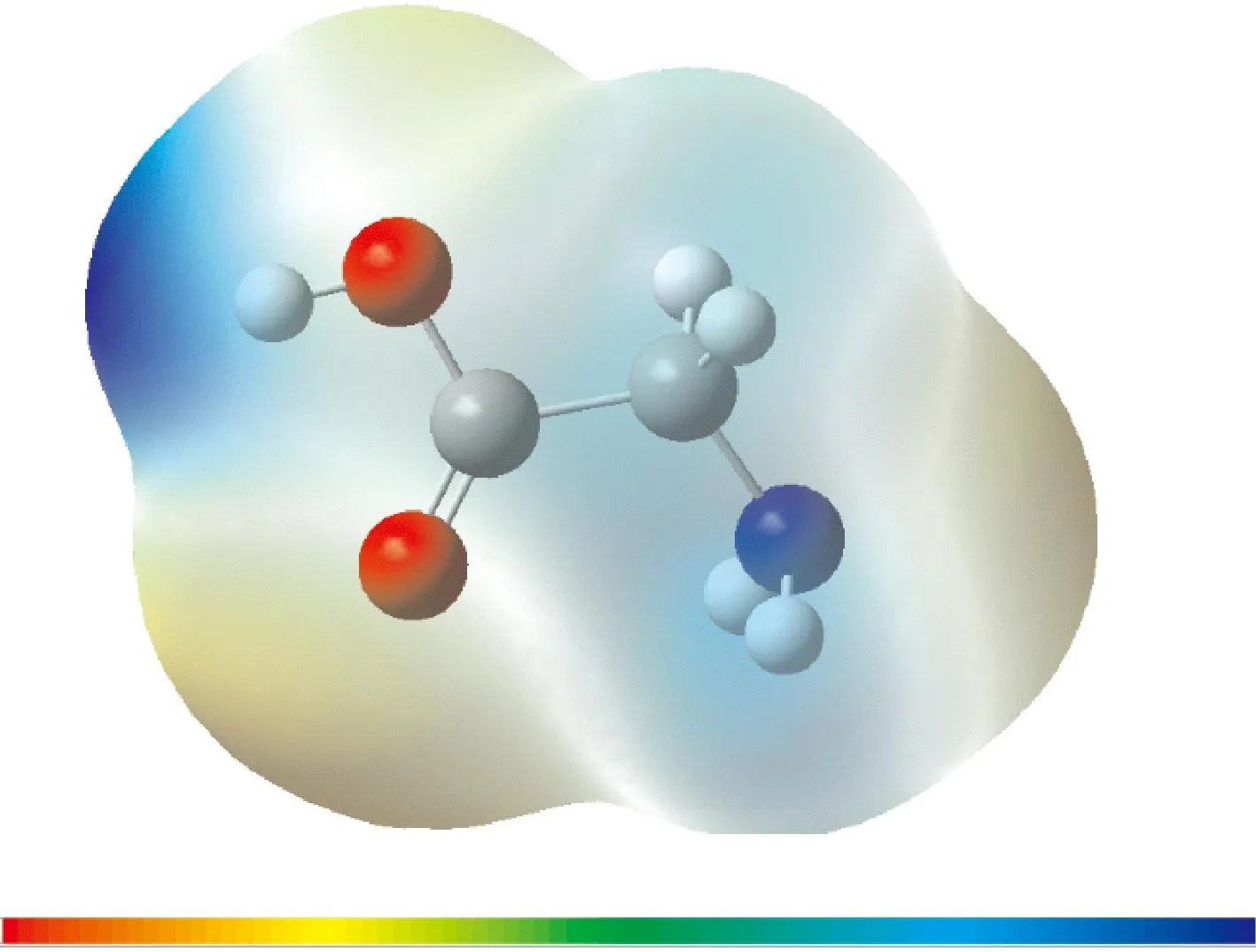
图1 甘氨酸分子的计算静电表面电位Fig. 1 The computed electrostatic surface potential of the glycine molecule
图2显示了优化后的甘氨酸单体和甘氨酸-H2O氢键复合物的基态结构. 根据对甘氨酸单体和氢键复合物的振动频率分析无虚频存在,所有优化的几何结构都已达到最低能量. 首先计算基态和激发态下甘氨酸单体分子和氢键复合物的重要结构数据,结果如表1所示. 在甘氨酸分子中7H和3O原子可与一个H2O分子形成分子间氢键,如图2(a)所示. 7H…8O和3O…9H氢键的长度分别为1.78378 Å和1.97986 Å. 值得注意的是,复合物结构(a)中化学键2O-7H和2C=3O的键长分别为0.98763 Å和1.21749 Å,与甘氨酸单体分子相比均被拉长. H2O分子中8O-9H的长度也因为氢键的形成而改变,增加了0.01214 Å. 在其他两个配合物中也观察到了这种现象. 产生这种现象的原因可能是氢键的形成,使得氢键供体原子的化学键伸长. 复合物结构(b)中氢键3O…10H和12H…8O的长度分别为1.92096 Å和2.14515 Å,但复合物结构(c)中相同位置的氢键长度不同,说明氢键的键长会受其他氢键的影响.

表1 氢键甘氨酸-H2O复合物和甘氨酸单体的键长(Å)
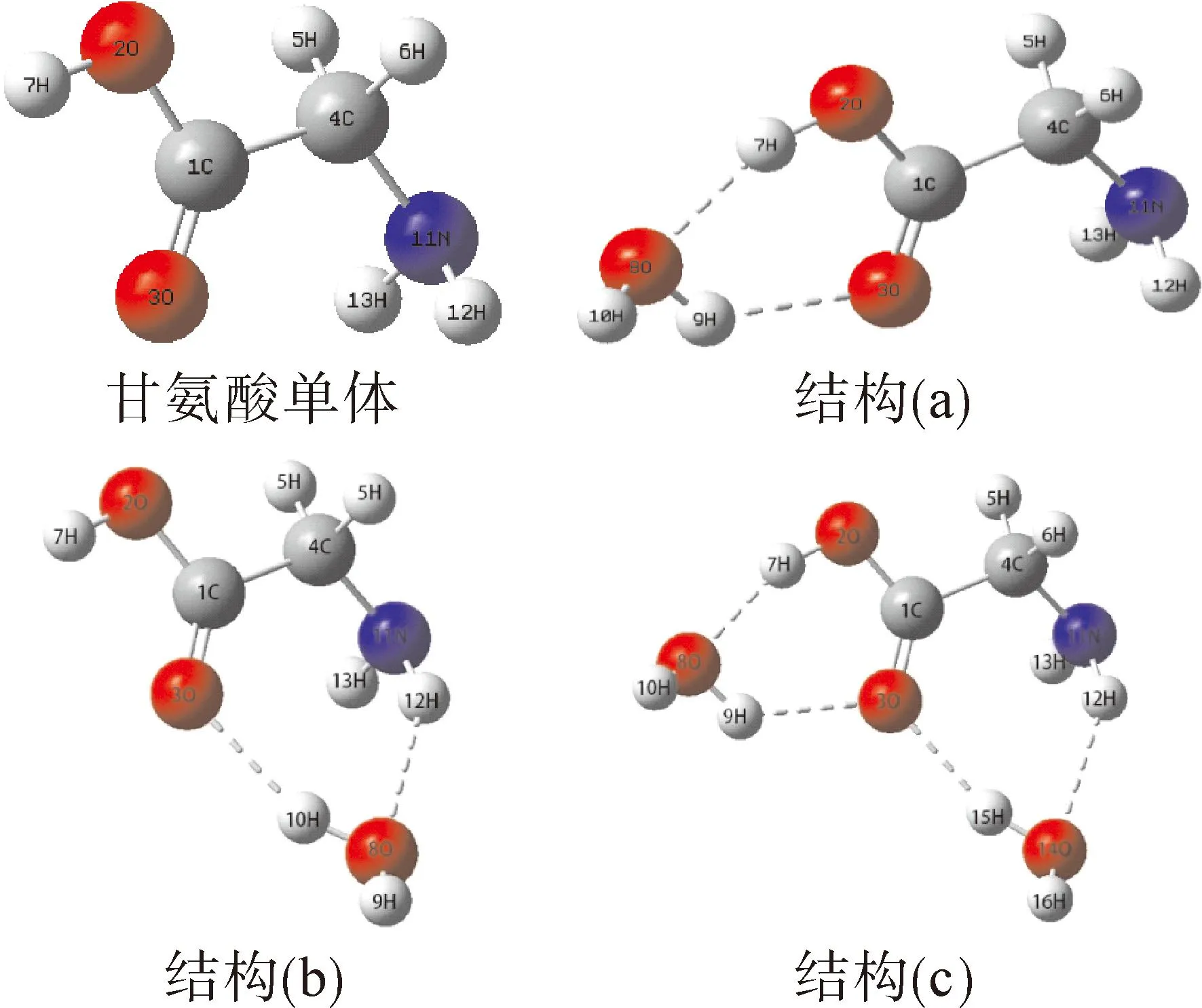
图2 甘氨酸单体和氢键甘氨酸-H2O复合物的几何结构Fig. 2 Geometric structures of glycine monomer and hydrogen-bonded glycine- H2O complexes
当上述四种结构处于激发态S1时,分子间氢键的键长都有不同程度的变化. 通过比较基态和激发态下分子间氢键长度的变化,可以看出相同结构中不同氢键有着不同的变化趋势. 当氢键复合物结构激发时,复合物结构(a)中氢键7H…8O长度从1.78378 Å增长到1.97633 Å,化学键强度减弱;分子间氢键3O…9H的长度从1.97986 Å缩短到1.74672 Å,强度增加. 在激发态下,与氢键相连的化学键长度变化趋势与氢键长度的变化趋势正好相反. 其中结构(a)的氢键7H…8O和化学键2O-7H的长度分别为1.78378 Å和0.98763 Å,而在激发态下,2O-7H键缩短,氢键7H…8O增长,两者呈现相反的变化趋势. 这种现象也可以在其他复合物中观察到,结合甘氨酸单体分子的变化可能是因为氢键的形成会影响化学键的键长和强度.
3.2 电荷分布
自然键轨道(NBO)方法常用于分析非共价相互作用[5,12]. 本文采用该方法计算电荷分布,计算结果如表2所示. 结构(a)中原子3O处电荷为-0.659,7H处电荷为0.514,对比甘氨酸单体处电荷数据,3O处负电荷增长0.054,7H处正电荷增长了0.031,可以发现分子间氢键的形成对电荷分布有影响,尤其是在形成氢键的位置上. 在H2O分子中,与氢键相连的H原子比其他位置的H原子携带更多的正电荷. 同样与氢键相连的7H和12H原子上的正电荷明显高于其他H原子,3O和2O原子上的负电荷明显高于其他O原子,甘氨酸与1C原子相连的O原子上正电荷也有增加的趋势. 通过对比可以发现激发态会影响大多数原子的电荷分布.

表2 甘氨酸单体和氢键配合物的自然键轨道(NBO)电荷分布.
3.3 自然键轨道和AIM分析
AIM分析是研究分子间相互作用和电子激发对甘氨酸与水分子间氢键影响的一种简单而有效的方法. 通过该方法获得三个重要的值:ρ电子密度;▽2ρ电子密度的拉普拉斯量;H氢键在键临界点处的电子能量密度[5,10]. Wiberg键级b通过NBO计算[5]. 计算得到的数据见表3.
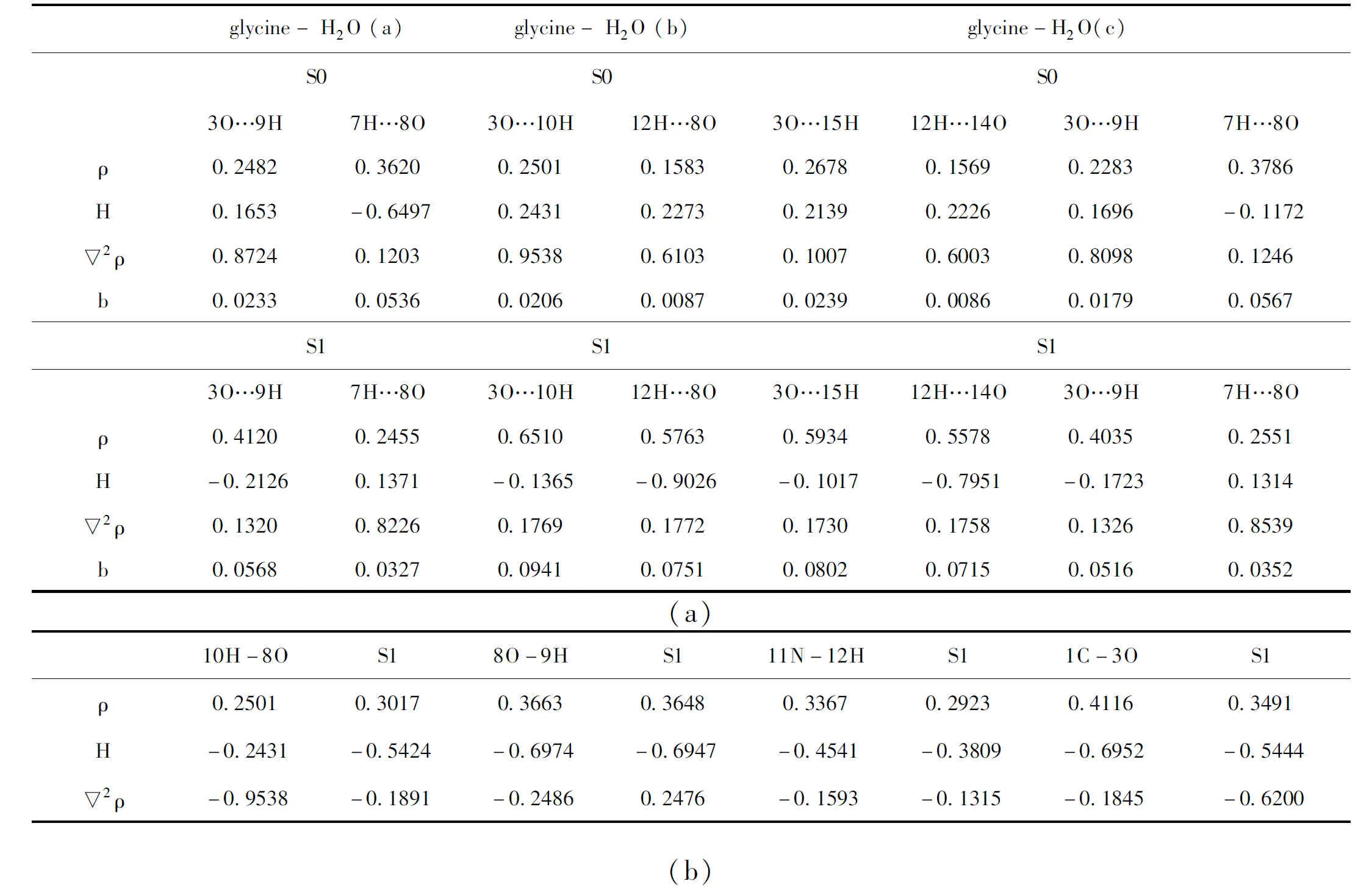
表3 复合物(a)中氢键的BCP处的Wiberg键级b,电子密度性质ρ,▽2ρ、H和甘氨酸水中的其他化学键的主要数据(b)
当氢键复合物被电子激发时,结构(a)中氢键3O…9H的ρ和键阶b增加,因此氢键3O…9H应该增强,这就解释了激发态下键长变短和频率红移的现象. 分子间氢键7H…8O削弱,相应的ρ和b对应减小. 在结构(b)中,激发态下的两个氢键都得到了加强,相应的ρ和b也对应增加. 这一规律也可以通过结构(c)中的分子间氢键的变化规律得到证实.
表3(b)展示了复合物结构(b)中主要化学键的数据变化. 在结构(a)的激发态下,化学键11N-12H和1C-30中b、▽2ρ和ρ发生了显著变化,这意味着2O-7H和1C-3O键被电子激发. 但是当复合物从基态激发到激发态时,H2O分子化学键8O-9H中b和▽2ρ,ρ和H的值几乎没有改变. 因此可以得出结论,水分子的化学键8O-9H部分受激发态影响较小.
图3展示了氢键复合物结构(b)的前沿分子轨道. 根据计算结果,氢键复合物的激发态对应从最高占据分子轨道(HOMO)到最低未占分子轨道(LUMO)的轨道跃迁,这里只描述了HOMO和LUMO轨道. 从图中可以发现电子云密度主要从氨基(-NH2)转移到羟基(-COOH),说明1C=O和11N-12H基团的电子密度的变化会影响氢键的形成. 同时,电子云密度的变化主要与水分子的甘氨酸部分和10H-9O部分有关,这与前面的观点是一致的.

图3 甘氨酸-H2O(b)氢键配合物基态HOMO和LUMOFig. 3 HOMO and LUMO of the hydrogen-bonded complexglycine-H2O (b)in the ground state
3.4 振动吸收光谱
为了更好地了解氢键在电子激发下的变化,计算了基态和激发态中上述三种结构的红外光谱变化. 通过比较单体分子和复合物的振动频率和振动类型,发现单体分子和复合物的振动谱中存在相似的振动峰. 氢键复合物的甘氨酸部分保留了单体分子的原有振动特性. 图4为甘氨酸-H2O复合物形成氢键后的各种新的振动峰.
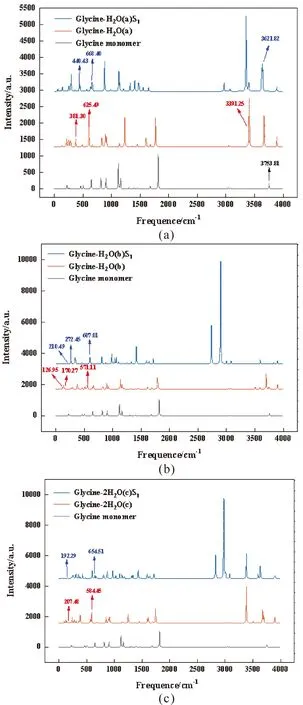
图4 单体和复合物的振动光谱:(a)结构a;(b)结构b;(c)结构cFig.4 Vibrational spectra of monomers and complexes:(a)structure a;(b)structure b;(c)structure c
在图4(a)甘氨酸单体的振动谱中,在3753.81 cm-1处出现了明显的振动峰值,为2O-7H的拉伸振动. 而在氢键复合物的振动谱中,振动红移至3391.25 cm-1. 当络合物被激发时,振动发生蓝移至3621.82 cm-1. 说明氢键的形成削弱了2O-7H键,激发态增强了2O-7H键,这与前面的结论相互印证. 在氢键络合物中,氢键3O…9H的摆动振动在381.30 cm-1和625.49 cm-1处,激发态下,该振动的分别蓝移至440.43 cm-1和668.40 cm-1处. 在红外图像中可以发现氢键3O…9H的增强,这与之前的结论一致
由图4(b)可知,在复合物的振动谱中,氢键3O…10H的拉伸振动和摇摆振动分别在170.27 cm-1和571.11 cm-1处. 激发态下,它们分别蓝移至272.45 cm-1和607.01 cm-1处. 在红外图像中可以识别出氢键3O…10H的增强. 同样氢键12H…8O的拉伸振动最初在126.95 cm-1处,当处于激发态时,频率变化至210.49 cm-1处. 从红外光谱上看,激发态增强,这与之前的结论相互印证.
在图4c中,结构(c)的氢键振动是复杂的. 例如氢键3O…9H和3O…15H的摇摆振动的振动峰值在584.45 cm-1处. 激发态下,该振动峰蓝移到654.51 cm-1处. 207.48 cm-1处的振动峰值是由氢键7H…8O和3O…9H的拉伸振动产生的,且在激发态下频率红移到192.29 cm-1处.
3.5 空穴电子与电子转移分析
电子转移发生在激发态下,是引起分子结构变化的重要原因之一. 表4中,在激发态S1下的三个结构中,甘氨酸中存在少量的电子转移至H2O中. 电子转移现象主要出现在甘氨酸片段中,其数量分别为0.97806、0.97016和0.93598. 激发态空穴和电子的变化如图4所示,电子的转移主要发生在甘氨酸内部.
表5显示了甘氨酸片段内的电子转移. 对比图5中的电子位置和空穴位置,在激发态下甘氨酸单体分子中的电子主要由NH2和CH2基团向羧基转移,净转移量分别为0.63273和0.10864. 在结构(a)中,电子主要从NH2基团转移到羧基上. 在结构(b)中,电子主要从NH2转移到羧基和CH2基团. 在结构(c)中,电子也主要从NH2基团转移到羧基和CH2. 氢键的产生会引起空穴电子的变化,相应的也会引起激发态电子转移的变化.

表5 分子内电子在组间转移,其中片段1代表COOH,片段2代表CH2基团,片段3代表NH2基团

图5 空穴电子分析:(a)甘氨酸单体;(b)结构a;(c)结构 b;(d)结构 c,其中蓝色代表空穴,绿色代表电子Fig. 5 Hole-electron analysis:(a)glycine;(b)structure a;(c)structure b;(d)structure c in which blue stands for hole and green stands for electron.
4 结 论
综上所述,本研究利用TD-DFT和DFT方法研究了分子间氢键的性质. 分子间氢键的形成可以延长为氢键提供原子的化学键的长度. 例如,随着分子间氢键的形成7H-2O、1C=3O和11N-12H的键长增加,这也影响了整个分子NBO的电荷分布. 其次,在激发态下,分子间氢键的强度会发生变化. 例如,在复合物结构(a)中,氢键7H…8O减弱,氢键3O…9H增强. 最后,根据单体分子和氢键络合物的红外光谱发现,当分子间氢键形成时,甘氨酸单体分子原有的振动峰值频率会发生位移,其运动方向与氢键强度的变化有关. 在激发态下,分子间氢键振动峰值的频率也会发生移动,移动的方向与氢键强度的变化有关. 在激发态下,H2O与甘氨酸分子之间存在少量的电子转移,主要转移量在甘氨酸分子中,在三种结构中,电子主要从NH2转移到COOH.
氨基酸与水的结合不仅在研究中很常见,在生命的形成和发展中也很常见. 通过探索氨基酸分子中H2O分子形成氢键的性质,可以更好地检验氨基酸在生物体内的存在状态. 同时,对其激发态变化的研究有望为今后分子间氢键的研究提供理参考.
猜你喜欢
上海计量测试(2022年4期)2022-02-01
汕头大学学报(自然科学版)(2020年4期)2020-12-14
考试周刊(2016年60期)2016-08-23
考试周刊(2016年48期)2016-06-29
中学生数理化·高二版(2016年6期)2016-05-14
人间(2015年11期)2016-01-09
原子与分子物理学报(2015年3期)2015-11-24
邵阳学院学报(自然科学版)(2015年2期)2015-06-05
中国洗涤用品工业(2015年7期)2015-02-28
原子与分子物理学报(2014年1期)2014-03-20
