
金属-有机框架材料的调控策略及其对典型重金属离子的吸附性能
2024-01-16 11:29许春树姚庆达梁永贤周华龙
化工进展 2023年12期
许春树,姚庆达,梁永贤,周华龙,4
(1 晋江市质量计量检测所,福建 泉州 362200;2 四川大学(石狮)先进高分子材料研究中心,福建 泉州 362700;3 兴业皮革科技股份有限公司,福建 泉州 362200;4 四川大学制革清洁技术国家工程实验室,四川 成都 610000)
未经处置的工业废水含有大量的有毒有害重金属和类重金属离子,若直接排放进入江河湖泊不仅污染地表水,导致水生动植物的死亡,还可通过食物链进入人体并富集,导致各种疾病和机能紊乱,最终对人体健康造成严重危害。金属冶炼、电镀电解、石油化工、制革工业、纺织印染等企业产生的废水是重金属废水的主要来源。目前处置重金属废水的常规方法包括化学沉降、膜处理、电化学转换、吸附、离子交换等[1-2]。在众多方法中,吸附被认为是最简单、有效的处置方法,这主要归功于吸附剂具有结构易于设计、合成制备简单、成本低廉、再生性强等优点[3]。基于木质素、纤维素、壳聚糖等天然高分子吸附剂具有资源丰富、无毒无害、生物相容性高等特点,但同时也存在吸附能力低、溶胀性强、物理力学性能差、选择性弱等问题[4-5],因此面对成分复杂、处置难度大的含重金属工业废水,亟须开发高稳定性和高吸附能力的新型吸附剂以提高综合治理效率。
金属-有机框架材料(metal-organic framework materials,MOFs)是由金属离子/团簇和有机配体通过配位键有序连接构筑的具有多孔结构的配位聚合物[6]。目前,基于溶液法、扩散法、水(溶剂)热法、固相反应法等制备技术已经趋于成熟,根据金属离子/团簇和有机配体的不同,可得到线形、四边形、四面体、八面体等MOFs。MOFs 具有较大的比表面积、特殊的孔隙结构、易于表面修饰等特点,加之金属离子/团簇、有机配体和MOFs 表面、孔隙中活性官能团都可以很好地螯合/交换重金属离子,使其有望成为水处理中的高效吸附剂。为了进一步提升MOFs 的使用性能,基于MOFs 的MOFs/离 子 液 体[7]、MOFs/氧 化 石 墨 烯[8]、MOFs/层状双金属氢氧化物[9]等MOFs 基复合材料已经受到广泛研究,复合材料表面的活性官能团可与重金属离子形成稳定的螯合复合物,对Hg(Ⅱ)、Ni(Ⅱ)、Cd(Ⅱ)、Pb(Ⅱ)、Cr(Ⅵ)等多种重金属离子表现出了极强的吸附性能。但是多元组分的引入可能会在处置过程中产生毒副产物[8]。有鉴于此,通过功能化修饰对MOFs 的拓扑结构进行调控,以提高MOFs的稳定性、吸附性是一种更为有效、方便的途径。
迄今为止,王崇臣等[10]、唐朝春等[11]和附青山等[12]已经对MOFs 的合成、发展及MOFs、MOFs 基复合材料对重金属离子的吸附性能进行相关综述,但未对MOFs及功能化MOFs与多孔性、水稳定性、表面性质、吸附性能等关系进行系统的阐述。有鉴于此,本文从构筑MOFs 的角度出发,根据MOFs的结构特点,重点梳理了MOFs的结构特性及调控策略,主要包括了MOFs的多孔性、表面活性、框架柔性、水稳定性、可扩展性、生物毒性和循环使用性,然后重点分析了MOFs 对Pb(Ⅱ)、Hg(Ⅱ)等典型阳离子型重金属离子和Cr(Ⅵ)、As(Ⅲ)/As(Ⅴ)等典型阴离子型含氧离子的吸附,最后对MOFs在重金属离子吸附领域的应用进行探究和展望。
1 MOFs结构特性的调控
MOFs 作为一种新型吸附剂,对重金属离子表现出较好的吸附性能。以均苯三甲酸为有机配体、Cu 为金属节点的HKUST-1 对Cd(Ⅱ)的吸附量可达219.05mg/g[13]。HKUST-1 也可有效地去除Ce(Ⅲ)[14]。通过溶剂热法合成的含有二苯甲酸结构的DUT-3对As(Ⅴ)的吸附能力达到33.91mg/g[15]。与传统沸石基材料不同的是,MOFs 的结构特性与组成MOFs的金属离子/团簇和有机配体的种类和连接方式有关,不同的定向组装拓扑结构赋予MOFs不同的结构特性。MOFs 的吸附能力高度依赖于MOFs 的结构特性,MOFs 的多孔性、表面活性和框架柔性是MOFs 作为吸附剂的必要条件,而水稳定性、可扩展性、生物毒性和循环使用性则是MOFs作为高性能水处理剂的重要性能指标。
1.1 多孔性
吸附首先考虑的因素便是孔道的尺寸和表面性质。通常情况下,MOFs 的有机配体具有较长的链长,有机配体与金属节点的连接构成了MOFs的孔隙,因此MOFs具有与沸石类似的永久多孔性。根据孔隙的大小可分为微孔(<2nm)、介孔(2~50nm)和大孔(>50nm)。从微观结构上看,在溶剂热法中,以Cr(Ⅲ)和对苯二甲酸构筑的MIL-101(Cr)具有典型的介孔结构,而使用2-氨基对苯二甲酸构筑的MIL-101(Cr)-NH2则为高度微孔,孔径为1.85nm[16]。若将使用HCl将MIL-101(Cr)-NH2进一步质子化,则可促进孔隙朝向更小的1.12nm转变[17]。而使用水热法构筑的MIL-101(Cr)-NH2则具有微孔/介孔二元孔隙结构,并可使用NaOH进一步微调MIL-101(Cr)-NH2的多孔性[18]。微孔结构的MOFs 表现出更强的水稳定性,而介孔、大孔MOFs表现出更强的吸附能力。
金属节点的替换也会改变MOFs的多孔性,2-氨基对苯二甲酸和Zr(Ⅳ)、Al(Ⅲ)、Cd(Ⅱ)构筑的UiO-66-NH2、MIL-53(Al)-NH2、Cd-BDC-NH2的孔隙分别为4.9nm、1.9nm、5.7nm[19]。有趣的是,配体的氨基化修饰赋予了MOFs 良好的荧光性能,并可应用于痕量重金属离子检测领域中[19-20]。通过增长有机配体的长度也可调整MOFs的孔径,Deng等[21]通过不同数量的亚苯基单元构筑了一维蜂窝状结构的MOF-74,重复的亚苯基单元可从1 个不断增长至11个,并实现孔隙在1.4~9.8nm的精确调控(图1),有机配体长度的增加可以有效延伸配位网络,亚苯基的活性还可进行侧基功能化从而提供更多的储存空间和吸附位点。利用有机配体的竞争配位可以调节MOFs 的孔径。Zhao 等[22]通过有机配体的竞争配位,在Zr(Ⅳ)与均苯三甲酸构筑MOF-808的过程中引入对苯二甲酸以调节孔径,当对苯二甲酸用量为40%时,MOFs 孔径从1.84nm 提升至2.22nm。Feng等[23]利用4-磺酸基苯甲酸与对苯二甲酸的竞争配位合成UiO-66,也得到了类似结论,磺酸基更强的配位能力使UiO-66 的孔径从1.1nm提升至2.0nm。竞争配位使得MOFs 中暴露了更多的活性羧基,MOFs 的负电性提升,与重金属离子的螯合能力也随之增强[22-23]。但是,MOFs 的自组装会倾向于形成致密的配位网络结构,孔径的增大往往伴随着结晶性差、稳定性差、结构网络穿插等问题。
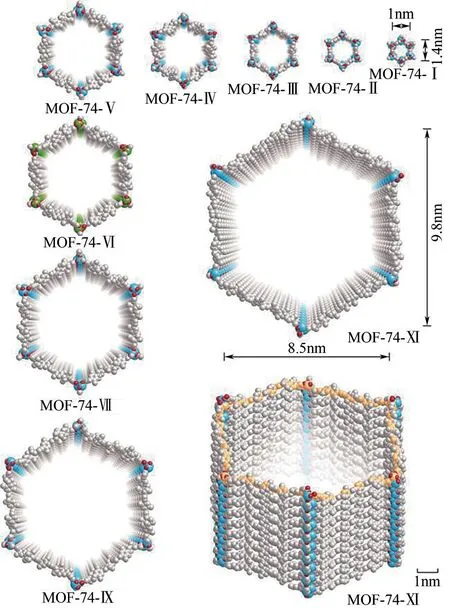
图1 不同亚苯基单元的多孔性MOF-74构筑策略[21]
具有二元孔隙结构的MOFs则可在保证框架完整性的前提下,改善MOFs孔隙的表面性质。在以丁二酸和Zr(Ⅳ)通过水热法构筑的高度介孔结构的Zr-SA中,对配体的巯基化修饰可生成一定量的微孔结构,并将MOFs 的比表面积从60m2/g 提升至513m2/g,同时提高了MOFs的结构稳定性[24]。Wang等[25]则是利用溶剂热法构筑Zr-SA-SH,N,N-二甲基甲酰胺的偶极矩较水更小,加速了MOFs 的成核,结构更为致密,孔径下降。此外,水热法所得Zr-SA-SH 的ζ 零点电位约为5.96eV,而溶剂热法所得MOFs在广泛的pH范围内显负电性,ζ零点电位仅为1.80eV[24-25]。使用均苯三甲酸和Fe(Ⅲ)可构筑的高度微孔MIL-100(Fe),Koo 等[26]使用磷酸对MIL-100(Fe)蚀刻,研究结果表明随着磷酸浓度的提升,MIL-100(Fe)结构中出现了2.43~18.44nm 的介孔,介孔结构的生成伴随着微孔结构的损失,其孔容也逐渐提升至1.1556cm3/g。此外,与盐酸相比,使用磷酸可以更好地保留MOFs的基本结构和特性,MOFs的框架并不会坍塌。
基于混合溶剂的微波辐照法也被证实是合成微孔/介孔二元孔隙结构MOFs的有效方法,通过调整二元溶剂的比例可以控制MOFs 的孔径,Yang 等[27]通过改变苯甲酸和N,N-二甲基甲酰胺的用量比例,使得高度微孔的Fe-MOF 结构中出现了5.9~12.2nm的介孔结构。与微孔结构Fe-MOF相比,介孔结构的出现促进了MOFs的生长成核,并提升其结构稳定性。利用有机配体网络的互相渗透,在构筑MOFs 时引入丙酸、乙酸等调节剂也可诱导MOFs形成微孔/介孔结构[28-29]。微孔/大孔二元结构MOFs具有更好的吸附性能。Ardila-Suárez 等[29]在溶剂热法合成MOF-808 的过程中引入表面活性剂,使用水/溶剂去除表面活性剂后则可获得微孔/大孔二元孔隙结构的MOFs,其孔径取决于表面活性剂的分子大小与浓度,其大孔尺寸可达172.0nm。Yang等[30]使用Cu(Ⅱ)和4,4′,4″-(1,3,5-三嗪-2,4,6-三基)三苯甲酸构筑介孔尺寸为31nm的MOFs,若在构筑过程中引入H2O2,还会在体系内出现尺寸为119nm的大孔,H2O2会破坏和蚀刻MOFs 晶体结构,孔径取决于H2O2的用量,但是过量的H2O2会破坏孔隙的规则分布和出现高度无序的介孔MOFs。自组装法是制备介孔/大孔MOFs的一种更简单的方法,通过改变初始配体的亲和力和在溶剂中的溶解度,可以控制MOFs 晶体生长的速率和各向异性,Zhang等[31]使用CuCl2和对苯二甲酸通过溶剂热法直接构筑了介孔/大孔二元孔隙结构的MOFs,其大孔孔隙可达150nm。
1.2 表面活性
未经修饰的MOFs可直接作为吸附剂应用于水处理中,但是构筑MOFs时,活性官能团倾向于与金属节点形成稳定的配位键,表面的活性官能团含量较低,吸附性能较差。提升MOFs的表面活性其一是破坏MOFs 的晶格网络,提升MOFs 的多孔性[16-31];其二是对MOFs 合成后修饰,巯基可与重金属离子形成稳定的配位作用,因此常用于MOFs的合成后修饰中。Chai 等[32]在水相中将UiO-66-NH2醛化,最后使用4,6-二氨基-2-巯基嘧啶改性制得巯基化UiO-66-SH[图2(a)]。Wang 等[33]则是使用巯基-1,3,4-噻二唑对醛化的UiO-66-NH2巯基化修饰[图2(b)],但是戊二醛醛化-巯基化改性接枝率较低,仅有约33%[32-33]。基于卤代烃的亲核反应具有选择性强、接枝率高的特点,Yuan 等[34]使用乙二硫醇利用—SH 与—Cl 的巯基化亲核反应制备了UiO-66-SH,乙二硫醇的亲核性极强,接枝率较乙二醇等亲核试剂更高,可达52.13%。从微观结构上看,三种巯基化修饰方法均保留了UiO-66 的八面体结构,并可通过静电相互-配位作用吸附Hg(Ⅱ)、Au(Ⅲ)和Fe(Ⅲ)[32-34]。
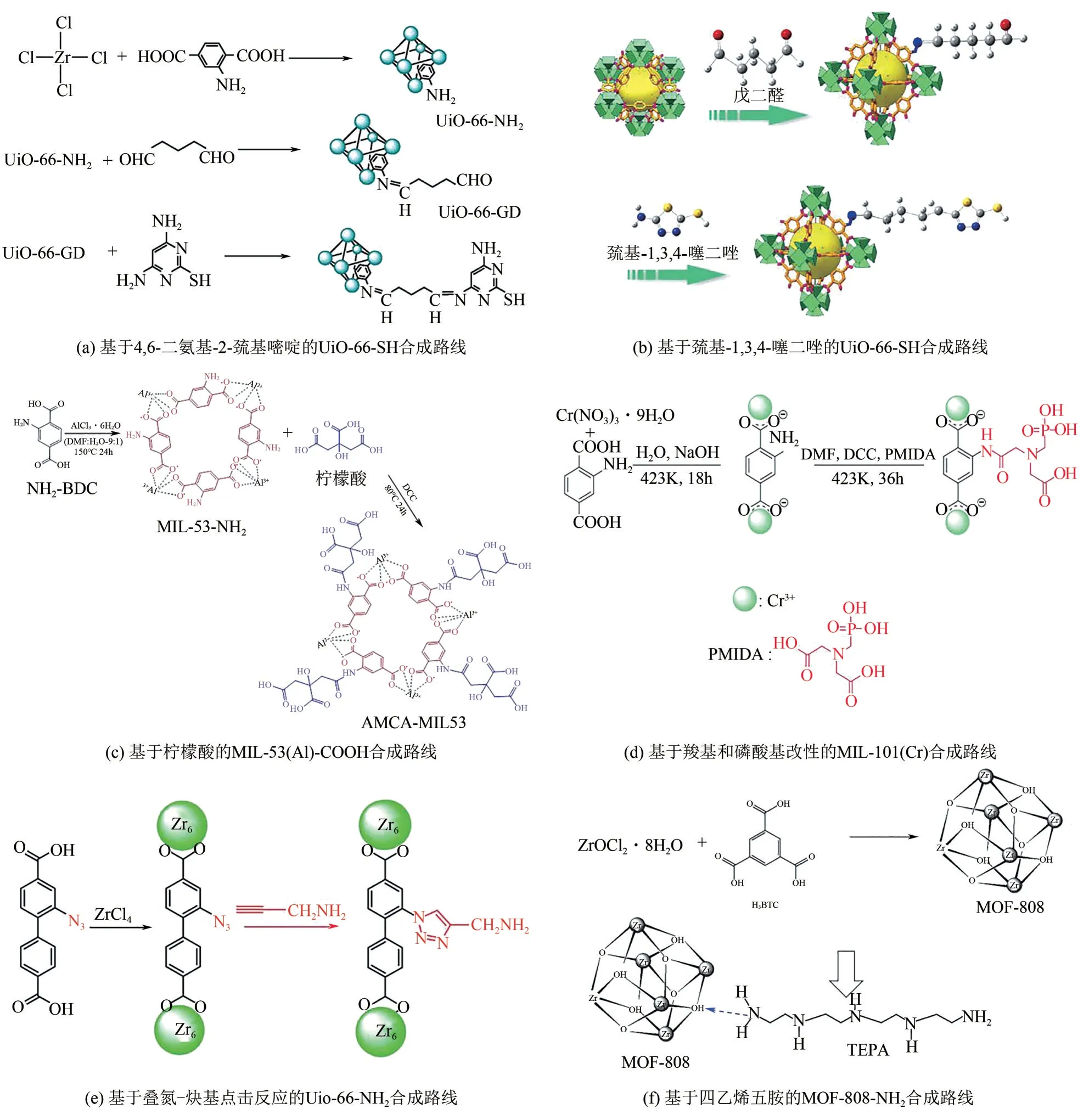
图2 基于合成后修饰的MOFs表面活性调控策略[32-33,35-36,41-42]
使用有机酸对MOFs表面改性是最常见的羧基化方法。有机酸的表面修饰使得MOFs在更宽泛的pH 范围内显负电性,Alqadami 等[35]在1,3-二环己基碳二亚胺催化下将柠檬酸接枝在MIL-53(Al)-NH2表面[图2(c)],柠檬酸的修饰使得MIL-53(Al)-NH2的ζ零点电位从8.0eV降低至4.1eV。Fu等[36]则是在MIL-101(Cr)-NH2表面羧基化改性的同时引入磺酸基,羧基和磺酸基的功能化修饰使得该MOFs在pH为2.0~6.0 的范围内高效吸附U(Ⅵ)[图2(d)]。Ji 等[37]使用强配位性的乙二胺四乙酸在水相中交换MOF-808结构上的甲酸分子,小分子甲酸被大分子乙二胺四乙酸交换后,其基础晶体结构并未发生改变,但对金属离子的螯合能力得到了显著提升,在海水淡化领域具有潜在应用价值。还可使用Eu(Ⅲ)和Ag(Ⅰ)进一步改性乙二胺四乙酸修饰MOF-808,不同配位数的金属离子交换破坏了MOFs 晶格的完整性,八面体形态完全消失,孔径增大,MOFs 暴露出更多的羧基,负电性得到进一步增强[38]。
基于氨基MOFs 的合成后修饰可以通过共价键、配位键、氢键和离子键等方式实现。Yi等[39]通过点击反应制备了基于氨基的共价键合成后修饰MOFs,首先通过溶剂热法合成UiO-66-N3,而后通过叠氮-炔基点击反应制备了氨基化UiO-66-NH2[图2(e)]。由于炔基端具有较为合适的链长,点击反应对微观结构的影响较常规化学接枝方法更小,比表面积仅下降约20%。Rani等[40]使用氨基酸替换HKUST-1 晶格中的均苯三甲酸,其中赖氨酸可最大程度保留HKUST-1 的结晶度和形态,而半胱氨酸则可将晶体结构转变为无定形。Jun 等[41]基于—OH 和—NH2的氢键相互作用设计了四乙烯五胺改性MOF-808[图2(f)]。Akbarian等[42]则是使用氯化胆碱季铵盐化1,4-苯基二异氰酸酯交联UiO-66-NH2制备了UiO-66-NH2R+。但这三种方法制得的氨基化MOFs 比表面积下降了约65%,孔容下降了约75%,这可能与包覆在MOFs 表面的有机物堵塞了孔隙有关[40-42]。
1.3 框架柔性
MOFs 的金属节点和有机配体形成的配位键以及有机配体自身存在的膨胀、收缩等动态变化赋予了MOFs框架柔性。对金属节点、有机配体的替换和有机配体的修饰可以有效控制MOFs 的框架柔性。MOFs在水相中的膨胀-收缩可以调整MOFs的孔隙,从而影响其水稳定性与吸附性能。
MIL-53(Cr)是一款具有极强框架柔性的MOFs,Gérard 等[43]通过溶剂热法制备的MIL-53(Cr)的体积为1.4405nm3,在水相中体积为1.0126nm3。两种状态下MIL-53(Cr)中O—Cr—O(88°~94°)和Cr—O—Cr(125°~132°)的键角差异不大,仅O—Cr—O 和O—Cr—Cr—O二面角的变化较大(177.5°和139.0°),这是MIL-53(Cr)极性基团和水分子之间的氢键作用引起的结构转变所致[图3(a)]。将MIL-53(Cr)金属节点替换为V(Ⅲ)后便丧失了框架柔性,但若替换为Fe(Ⅲ)、Al(Ⅲ)仍表现出较好的框架柔性。有趣的是,MIL-53(Fe)的框架柔性较MIL-53(Al)更差,这可能与金属节点未配对电子的相互作用会提升框架的稳定性有关[43-44]。若改变合成条件,使用水热法构筑MIL-88B(Fe),则可恢复框架的柔性[44-45]。进一步地,将对苯二甲酸分别替换为富马酸、2,6-萘二羧酸、4,4′-联苯二羧酸构筑MIL-88A(Fe)、MIL-88C(Fe)、MIL-88D(Fe)也具有较好的框架柔性[45-46]。其中MIL-88A(Fe)在水相中体积变化可达85%,孔径可从0.926nm变化至1.387nm[46]。
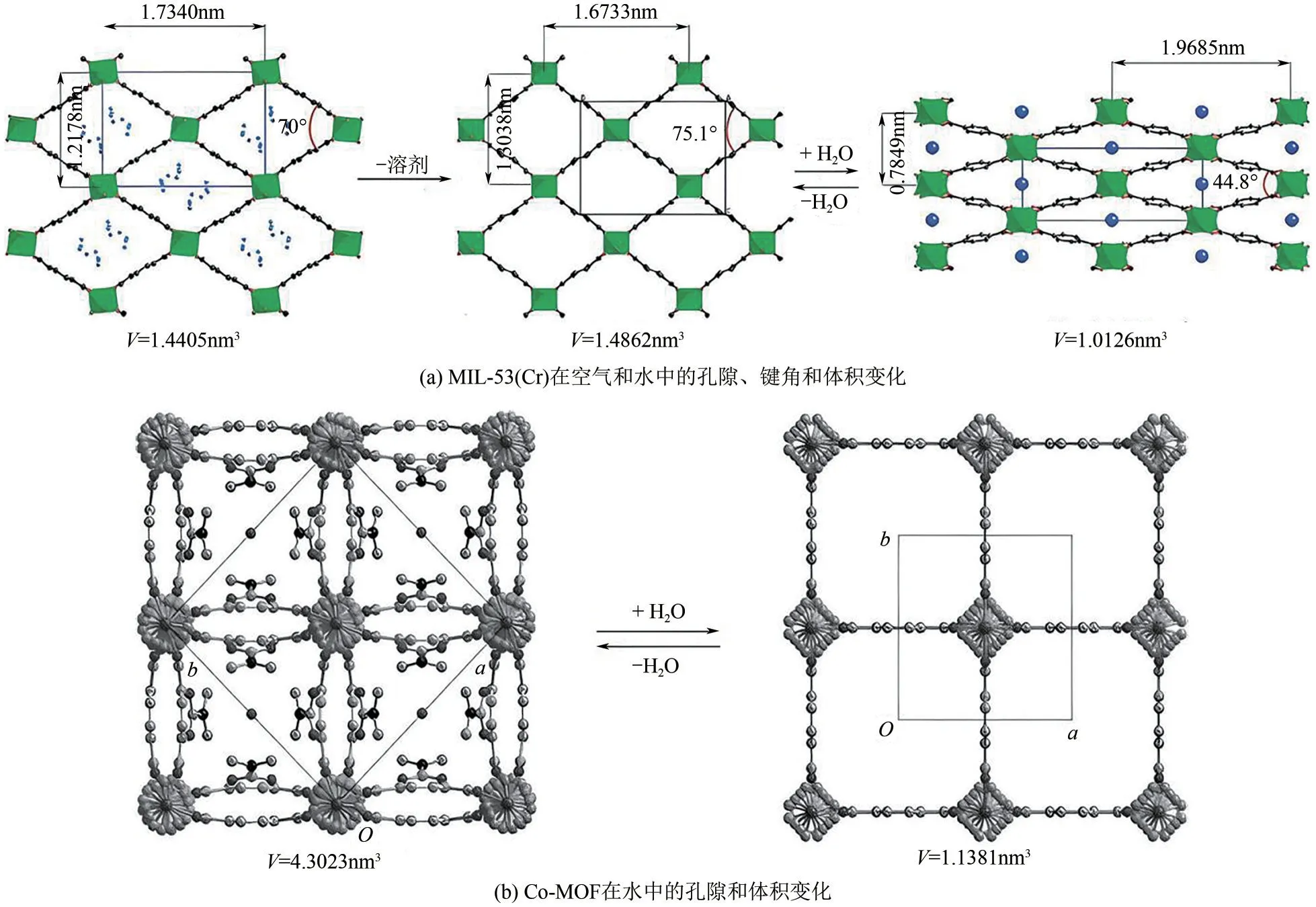
图3 MOFs的框架柔性[43,47]
Wang等[47]使用Co(Ⅱ)和对苯二甲酸、1,4-乙烯哌嗪构筑的高度介孔Co-MOF被证实具有极好的框架柔性,在水相中其介孔尺寸和数量均有所提升,在晶格中的表现为:空气中苯与羧基的二面角为0°,而暴露在水中时二面角变为8.3°。此外,在水相中还可观察到Co-MOF中有个三维互联的孔隙,沿着c轴的通道尺寸为0.76nm×0.76nm,沿着a、b轴的孔隙通道为0.51nm×0.37nm[图3(b)]。但将金属节点由Co(Ⅱ)替换为Cu(Ⅱ)后,Cu-MOF 表现了极强的刚性,框架柔性几乎完全丧失,在水相中难以改变基础孔隙结构[48]。2,6-萘二醚较对苯二甲酸具有更好的水分子亲和力,MOFs的基本框架更易膨胀-收缩,使用2,6-萘二醚、1,4-乙烯哌嗪构筑的DUT-8(Co)和DUT-8(Cu)也得到了类似结论,DUT-8(Co)具有更好的框架柔性,而DUT-8(Cu)则具有更强的刚性[49]。
在MOFs中,有机配体可充当为连接金属节点的桥梁,因此有机配体的几何构型、功能性都会影响MOFs的框架柔性。在有机配体的侧基中引入活性官能团是一种调节MOFs框架柔性的简单方法,在Zr(Ⅳ)和富马酸构筑的MOF-801中,甲基化富马酸表现出更高的孔隙率,但是其比表面积(756.2m2/g)较MOF-801(864.0m2/g)小得多,这是因为孔隙率的提高是建立在甲基化MOF-801 的框架柔性差的基础上的[50]。Cheang 等[51]使用均苯三甲酸和均苯四甲酸构筑的MIL-101(Fe)-COOH 和MIL-101(Fe)-(COOH)2孔隙率较MIL-101(Fe)也有明显提升,这是因为“笨重”的羧基侧链会限制或抑制MOFs的框架柔性,且侧基越大,框架柔性越差;而使用含N的有机配体(如吡嗪、4,4′-联吡啶、哌嗪等)或对有机配体氨基化修饰则有利于提升MOFs的框架柔性[16,47]。
1.4 水稳定性
MOFs 的水稳定性主要指其在酸、碱、盐溶液中保持框架结构和框架柔性的能力。应用于水处理的MOFs 如UiO-66、MIL-53、MIL-101 和MOF-808 等具有较好的耐酸稳定性,而ZIF-8 和ZIF-67等则是具有较好的耐碱稳定性,这种差异可以通过软硬酸碱理论进行解释[11]。调节MOFs 的结构参数也可改善MOFs的水稳定性,如图4所示。
图4 MOFs的水稳定性调控策略
水作为无机配体,可通过配体交换破坏MOFs的结构,因此提高MOFs 的疏水性可以提升MOFs的稳定性。以咪唑类为配体的ZIF 系列MOFs(ZIF-1~ZIF-12)中,由Zn(Ⅱ)与2-甲基咪唑配位形成的ZIF-8 具有最强的水稳定性,可在50℃的苯、甲醇、水和NaOH 水溶液中稳定存在7 天,这可能是因为2-甲基咪唑上含有疏水的—CH3所致[52-53]。在另一个通过对苯二甲酸和Zn(Ⅱ)构筑的Zn-MOF中也观察到类似现象,四甲基对苯二甲酸所构筑的Zn-MOF较硝基、羟基、溴基对苯二甲酸具有更强的水稳定性[54]。提高金属节点-有机配体的配位键强度,降低水与有机配体的替换也有利于提升MOFs的水稳定性,如使用磷酸、羧酸、磺酸等侧基功能化有机配体构筑MOFs显示出了更强的水稳定性[51]。金属离子的掺杂改性也会提升MOFs的水稳定性,Qin等[55]使用Tb(Ⅲ)替换Cd-MOF的金属节点,其疏水性得到显著增强,该MOFs在碱溶液中表现了极强的稳定性,并可在水相中稳定存在半年以上。Xia 等[56]发现Eu(Ⅲ)对金属节点的替换可进一步提升UiO-66 的水稳定性,该MOFs 可在pH范围为4~11的水溶液中稳定存在,并高效监测痕量的Fe(Ⅲ)和Al(Ⅲ)。
通常情况下,MOFs 上的缺陷可作为高活性位点用于吸附污染物,缺陷的产生会一定程度上降低MOFs 的水稳定性[19-23]。从结构调控的角度上看,提高MOFs水稳定性还可提高金属离子/团簇或有机配体的配位数。以UiO-66为例,每个Zr6O6金属节点都与12个对苯二甲酸配体相连接[23]。在另一个具有极强水稳定性的PCN-250中也发现了Fe3的12个连接节点[57]。Cr(Ⅲ)具有动力学上的惰性和高电荷/半径比,以Cr(Ⅲ)为金属节点的MIL-53(Cr)、MIL-100(Cr)和MIL-101(Cr)等均表现出了较强的水稳定性[43-45]。Yang 等[58]构 筑 了 一 种 基 于Cr(Ⅲ)的9 配 位CPM-243,该MOFs可在pH<0至pH>14的大约16个pH单位的极端条件下稳定存在48h。但是高配位数意味着MOFs的活性位点损失,吸附性能下降。
与柔性配体相比,具有较强刚性的有机配体可提高MOFs的变形能垒,并保持其基本构型。与长链的有机配体相比,短链的配体具有更强的刚性,这可能是因为在桥联配位MOFs的构成中,短链有机配体会弯曲成更大的角度,从而增加MOFs水解的活化能[45]。例如,UiO-66和UiO-67都是以Zr(Ⅳ)构筑的MOFs,UiO-66 的有机配体为对苯二甲酸,较UiO-67的4,4′-联苯二甲酸配体刚性更强,因此UiO-66 具有更强的水稳定性[59]。固定柔性的有机配体也有利于提升MOFs的水稳定性[60]。
有机配体的官能团修饰也会影响MOFs的水稳定性,侧基功能化在影响有机配体刚性的同时也会影响其对称性。羧基、磺酸基等大分子官能团的过度引入会使配体的结构发生扭曲,配体的过度变形终将导致MOFs 稳定性的下降[22-23]。此外,官能团的吸电子和给电子效应也会改变配体的抗氧化性能,从而影响MOFs的水稳定性。吸电子官能团如巯基、羧基、卤素、硝基等可以减少MOFs结构上的游离电子,通过提升MOFs的抗氧化能力提升其水稳定性;而给电子官能团如氨基、羟基、烷(氧)基等会使MOFs的基本框架中出现富电子区,使其容易受到氧自由基的攻击而使配体被氧化分解[53-54]。
1.5 可扩展性
绝大多数MOFs 的合成反应都是在水(溶剂)中进行的,水(溶剂)的扩散促进了MOFs的成核与结晶,并可充当构筑多孔结构的模板剂。Oh 等[61]在一个可扩展体系中将反应体积从10mL 扩大至225mL,连续制备了MOF-74、MOF-174 和MOF-184,并可保证MOFs的基本结构特性。Velasco等[62]通过溶剂热法实现了6L级别的Y-MOF合成,产量可达100g,但是反应时间需要60h。水(溶剂)热法反应条件较为苛刻,通过优化反应条件,减少水(溶剂)法的压力、反应温度和时间也可以合成MOFs。Li等[63]等在383K温度下合成高质量的MIL-88A(Fe),使MIL-88A(Fe)的合成时间从传统合成的24h 缩短到0.5h,并可保证极高的比表面积。基于微波辐照的溶剂热法已被证实也可以大幅度减少反应时间[27]。但是水(溶剂)热法往往需要对多种溶剂进行反复筛选和实验,复杂的合成程序和有毒有害的溶剂限制了MOFs的大规模生产。
目前,在合成MOFs构筑过程中减少溶剂的使用成为新的研究热点。Leon等[64]通过机械球磨法将AlCl3、InCl3和有机配体反应合成MOFs,此法无需溶剂且可灵活控制反应规模,易于大规模制备MOFs。但是机械球磨法对MOFs 的物理力学性能有较高要求,且机械球磨法所得的MOFs孔隙率较低,难以构筑介孔、大孔MOFs。Cao 等[65]在含有2-甲基咪唑的乙醇和Co(NO3)2的水混合电解液中,采用金属电极作为阳极,在100℃和1~20mA/cm2的电流密度下,通过化学气相沉积法在溶液中和电极上制备了ZIF-67 晶体。通过电化学合成法所制备的MOFs具有极好的框架柔性,但是暂无通过此法制备用于水处理的MOFs的报道。尽管这些研究预测了MOFs的可扩展性,但在大规模生产方面仍存在很大差距。实际上,基于配位键构筑的MOFs框架结构极易受到外部化学环境的影响,MOFs 功能化可以改善其稳定性和框架柔性,但也会增加生产制造成本。
1.6 生物毒性
MOFs 作为水处理吸附剂,其生物毒性是一项重要的评价指标,但在文献中却鲜见MOFs的毒理学数据报道。常用于水处理的UiO-66和UiO-67显示出极弱的细胞毒性。有趣的是,UiO-66-NH2和UiO-66 具有相当的生物毒性,这种情况在MIL-125(Ti)和MIL-125(Ti)-NH2中也可被观察到,说明基于氨基的侧基功能化对MOFs的生物毒性影响不大[66-67]。使用不同的金属离子和2,5-二羟基对苯二甲酸构筑的系列MOF-74 显示出不同的生物毒性,MOF-74(Co)、MOF-74(Ni)和MOF-74(Mg)均具有较弱的细胞毒性,而细胞在200μg/mL 的MOF-74(Cu)、MOF-74(Mn)和MOF-74(Zn)暴露24h 后细胞活力仅剩(20.9±2.7)%、(18.3±3.0)%和(38.8±3.6)%[67-68]。以Zn(Ⅱ)为金属节点,分别与苯并咪唑、2-甲基咪唑构筑的ZIF-7、ZIF-8 均显示出了一定的生物毒性[53]。但同样是以2-甲基咪唑为有机配体,金属节点替换为Co(Ⅱ)构筑的ZIF-67 却有较强的细胞毒性,这是因为Co(Ⅱ)可诱导自由基的产生并破坏细胞膜的结构,引发红细胞的溶血作用,从而破坏血红蛋白的结构和功能。自由基的产生又会进一步地破坏ZIF-67 的有机配体,破坏了ZIF-67 的结构,释放出更多有毒的Co(Ⅱ)[69]。
在较低的剂量下,MIL-100(Fe)和MIL-101(Fe)均显示了较弱的细胞毒性,但MOFs 用量超过200μg/mL 时,24h 后细胞活力仅余(53.2±1.0)%和(41.8±7.3)%[67]。另外两种Fe(Ⅲ)基MIL-88A、MIL-88B 的细胞毒性也可被观察到,这可能是因为Fe-MOF 更易在生物体内形成活性氧,并通过炎症诱导细胞程序性死亡[70]。Cu(Ⅱ)分别和均苯三甲酸、3,3′,5,5′-联苯四甲酸、5,10,15,20-四(4-羧基苯基)卟啉构筑的HKUST-1、NOTT-100、Cu-TCPP均显示出了较强的生物毒性,但细胞毒性较Cu 自由基低,其中HKUST-1 对斑马鱼胚胎和成体的半抑制浓度IC50 分别为2.13mg/L 和1.50mg/L[71]。同样以5,10,15,20-四(4-羧基苯基)卟啉为有机配体,与Hf(Ⅳ)构筑的Hf-TCPP 却显示出了较弱的生物毒性,细胞在5μg/mL 的Hf-TPCC 暴露24h 活性不会发生改变,且在4μg/mL 的Hf-TPCC 中暴露120h,活性仅下降约20%[66]。
总的来说,水相中MOFs的生物毒性主要来自于MOFs结构不稳定或框架柔性过高,导致的部分结构在水体中水解,释放了重金属离子,重金属离子所产生毒性抑制了细胞活性。部分典型MOFs的生物毒性如表1所示。
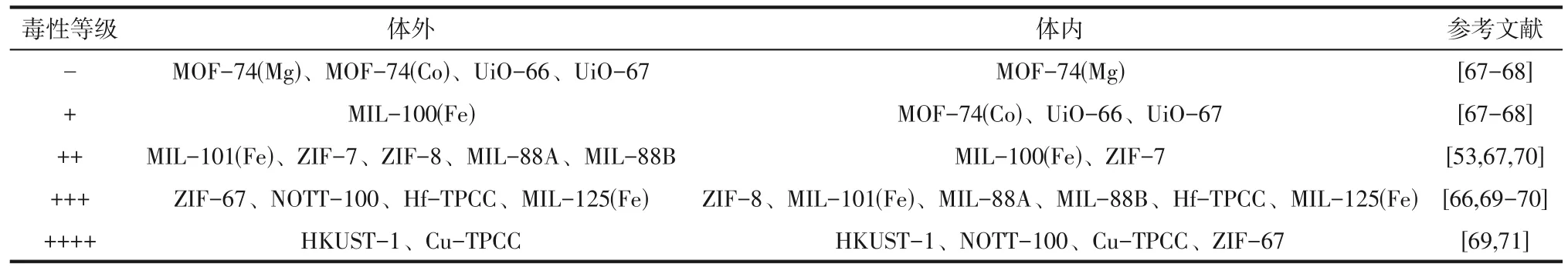
表1 部分典型MOFs的生物毒性总结
1.7 循环使用性
为了实现MOFs在实际水处理中的应用,要求MOFs 有较好的循环使用性。因此,在大多数研究中,都有对MOFs的循环使用性测试。通常情况下,MOFs 在被重复使用几次后仍然具有良好的吸附活性,Fu 等[36]研究了羧酸/磷酸功能化MIL-101(Cr)对U(Ⅵ)的吸附性能,并使用不同浓度的HNO3进行解吸附,研究结果表明0.3mol/L 的HNO3效果最好,一次吸附-解吸循环可恢复95.19%的吸附性能,四次循环后仍可保持66.95% 的初始饱和吸附量。Alqadami 等[35]使用柠檬酸合成后修饰MIL-53(Al)-NH2吸附水体中的Pb(Ⅱ),在吸附完成后,分别使用0.01mol/L的HCl、HNO3和H2SO4进行解吸。研究结果表明,HCl 作为洗脱剂可以最大保留MOFs 的吸附活性,这可能是因为Cl-半径最小、结合能力最强,更容易进入MOFs 的孔隙中与Pb(Ⅱ)结合并解吸。除强酸外,中强酸也可作为MOFs 的洗脱剂,Lyu等[20]研究了MIL-101(Cr)-NH2对重金属离子的吸附性能,在吸附-解吸过程中,使用0.1mmol/L的柠檬酸作为洗脱剂,经过6 次吸附-解吸循环后仍可保留对Fe(Ⅲ)、Cu(Ⅱ)和Pb(Ⅱ)约88.1%、78.8%和76.9%的最大饱和吸附量。
在MOFs 的吸附-解吸循环中,造成吸附性能下降的主要原因是MOFs 的晶体结构遭到破坏。Li等[72]研究了Y-MOF对Sb(Ⅴ)的吸附性能和循环使用性能,研究结果表明,洗脱剂对Sb(Ⅴ)的解吸能力按HCl、NaOH、Na2CO3、NaCl、Na2SO4和NaNO3递减,但是使用强酸或强碱会影响Y-MOF 的晶体结构,例如HCl 中的Cl-会替换Y-MOF 的有机配体,NaOH 会使部分Y-MOF 转化为YOx。以NaCl 作为洗脱剂时,Y-MOF 的循环使用性最佳,经过五个吸附-解吸循环后,仍可恢复68%的吸附效率。此外,有机溶剂也可在一定程度上缓解洗脱剂对MOFs结构的破坏,Wang等[32]使用巯基功能化UiO-66吸附回收水体中的Au(Ⅲ),再生实验中使用10%的硫脲和5%的盐酸作为洗脱剂,研究结果表明,UiO-66-SH 具有较好的循环使用性能,三次循环后饱和吸附量仅从147.39mg/g 下降至130.50mg/g,仍可保持88.50%的吸附效率。总的来说,MOFs的循环使用性与其水稳定性有关。通常情况下,MOFs的水稳定性越强,其循环使用性也越佳。
2 MOFs 对正电性重金属离子的吸附性能
2.1 铅(Pb)
Pb 是一种污染性较大的重金属,相对原子质量为207.2,Pb 的毒性作用对骨髓造血系统和神经系统损害较为严重。Roy 等[73]通过溶剂热法构筑了ZIF-8并用于吸附Pb(Ⅱ),研究结果表明,ZIF-8对Pb(Ⅱ)的吸附在溶液呈弱酸性至中性时最佳,这是因为当pH 较低时,Pb(Ⅱ)主要以Pb2+的形式存在,但此时ZIF-8 显正电性且稳定性较差;随着pH 的上升,Pb2+逐渐向Pb(OH)+和Pb3(OH)42+转变,ZIF-8表面携带电荷也逐渐转化为负电荷,吸附量逐渐提升;在碱性环境中Pb(Ⅱ)主要以Pb(OH)2、Pb(OH)3-等沉淀、凝胶的形式存在,此时的吸附作用转为物理吸附[图5(a)]。
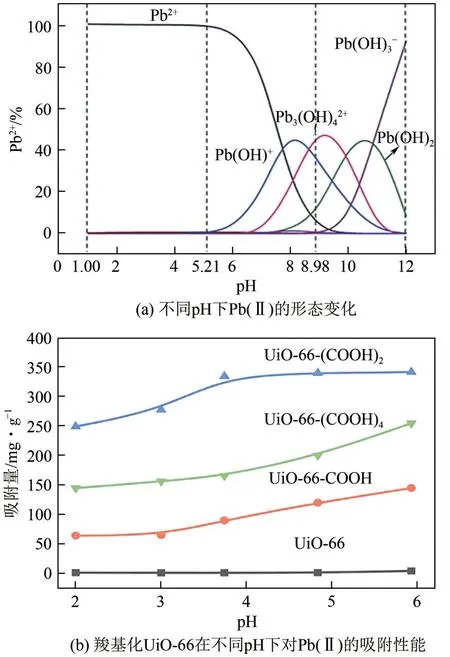
图5 pH对Pb(Ⅱ)形态及MOFs吸附性能的影响[73-74]
羧基化改性可以降低MOFs的等电点,使其在较低的pH 下也可以高效吸附Pb(Ⅱ),Zhao 等[74]使用均苯四甲酸构筑了UiO-66-(COOH)2,该MOFs具有极强的耐酸稳定性,在pH为2时仍可对Pb(Ⅱ)保持超过200mg/g的吸附量[图5(b)]。羧基化改性还提升了UiO-66-(COOH)2的循环使用性能,在4 次吸附-解吸循环后,仍可恢复约85%的吸附性能。在pH较低时,Pb(Ⅱ)主要以粒径较大的水合离子形式存在,为了进一步提升MOFs在较低pH下对Pb(Ⅱ)的吸附性能,Li 等[75]使用酒石酸对UiO-66-NH2合成后修饰,研究结果表明,酒石酸对2-氨基对苯二甲酸的交换破坏了UiO-66-NH2的微孔结构,MOFs 上出现了26~50nm 的介孔和50~120nm 的大孔,超大的孔隙结构不仅可以促进Pb(Ⅱ)在MOFs内部的扩散,还提升了MOFs 的刚性,对Pb(Ⅱ)表现出了186.14mg/g的最大饱和吸附。此外,通过合成后修饰在MOFs表面接枝负电性的有机物也有利于提升对Pb(Ⅱ)的吸附性能和循环使用性能[76]。
在最新的一项研究中,Zaman 等[77]利用环氧基与氨基的SN2亲核反应的原理将甲基丙烯酸缩水甘油酯接枝在UiO-66 的表面上,长链的聚丙烯酸链段可以提升MOFs 上的电子云密度,通过N—O 协同作用高效吸附Pb(Ⅱ)。但是甲基丙烯酸缩水甘油酯接枝后,UiO-66-NH2的孔隙率和比表面积出现了一定下降,这与有机物堵塞MOFs 的孔隙有关。在聚合物高分子链段上构筑MOFs 可以有效提升MOFs 的比表面积并改善其孔隙结构,提升MOFs与聚合物链段的相容性,这种构筑方法可以有效提升MOFs 的吸附位点,但是MOFs 在水溶液中的稳定性受到一定损失[77-78]。
2.2 汞(Hg)
Hg 属于典型的重金属原子,相对原子质量为200.6,Hg 易与蛋白质和酶结合,抑制其生物活性并可能致其失活。Hasankola等[79]使用5,10,15,20-四(4-羧基苯基)卟啉和Zr(Ⅳ)构筑了富含羧基的Zr-TCPP,研究结果表明当pH 较低时,Hg(Ⅱ)在Zr-TCPP上的吸附主要依赖阳离子-π电子云间的相互作用和离子交换,随着pH 的增加,相互作用逐渐转化为静电相互作用和配位作用,当pH超过Hg(Ⅱ)的沉淀值时,吸附作用转变为Hg(OH)2在Zr-TCPP孔隙中沉积与填充的物理吸附。
改变MOFs 的多孔性有利于提升与Hg(Ⅱ)的相互作用,Alshorifi 等[80]在构筑MIL-101(Fe)的过程中引入配位数不同的Co(Ⅱ),当Co(Ⅱ)和Fe(Ⅲ)摩尔比为1∶1 时,MOFs 的比表面积和孔隙达到最大,此时MOFs 中暴露出的羧基也最多,对Hg(Ⅱ)的吸附量可达312.97mg/g。在另一项研究中,Somayeh等[81]发现,在溶剂热法制备MOFs 的过程中引入少量的疏水链段即可通过限制聚合和定向组装,构筑有序微孔结构的疏水性TMU-59,疏水链段的引入提升了MOFs的水稳定性,抑制了Mn(Ⅱ)、Zn(Ⅱ)、Al(Ⅲ)、Fe(Ⅲ)等离子与TMU-59 金属节点的交换,Hg(Ⅱ)在TMU-59 上的吸附主要发生离子交换和配位作用,其最大吸附量可达1500mg/g。在最新的一项研究中,Ji等[82]通过配体交换的合成后修饰方法,使用α-巯基乙酸交换MOF-808结构中的甲酸制备了MOF-808-SH,MOF-808-SH上的活性羧基可以在较低的pH 下螯合Hg2+,而巯基则可在较高pH 下与Hg(OH)+、Hg(OH)2形成稳定的不带电荷的配合物—S—Hg—OH。此外,该团队使用0.1mol/L的硫脲和0.01mol/L 的HCl 再生MOF-808-SH,在6次吸附-解吸循环后仍可保持99%的最初饱和吸附量,从XRD图谱中也可发现重复使用的MOF-808-SH具有与原始样品相同的特征峰。
在实际应用中,Hg(Ⅱ)常出现于高盐废水中,因此对MOFs 的耐盐和耐水解性能有了更高的要求。Zhao 等[83]发现在高盐废水中,UiO-66-NH2表现出了较UiO-66 更好的吸附性能,这是因为高盐废水中Hg(Ⅱ)通常以HgCl2、HgCl3-和HgCl42-等形式存在,与表面携带有一定正电荷的UiO-66-NH2具有更强的相互作用,结果如图6所示。此外,在高盐废水中,MOFs 孔隙越大,越有利于Hg(Ⅱ)的扩散与结合,这是因为Cl-与Hg2+的配位化合物具有较大的分子尺寸。但即便是通过合成后修饰构筑氨基化MOFs,仍无法避免破坏MOFs结构的完整性,UiO-66-NH2存在的水中稳定性受到了一定损失。构筑含氮杂环MOFs或使用交联剂封闭氨基可有效解决此问题,但是交联剂的使用会封闭MOFs的孔隙,造成吸附性能的下降;而含氮杂环的MOFs则存在生产制备成本过高的问题[84]。部分MOFs 对典型的阳离子型重金属离子Pb(Ⅱ)和Hg(Ⅱ)的吸附性能见表2。
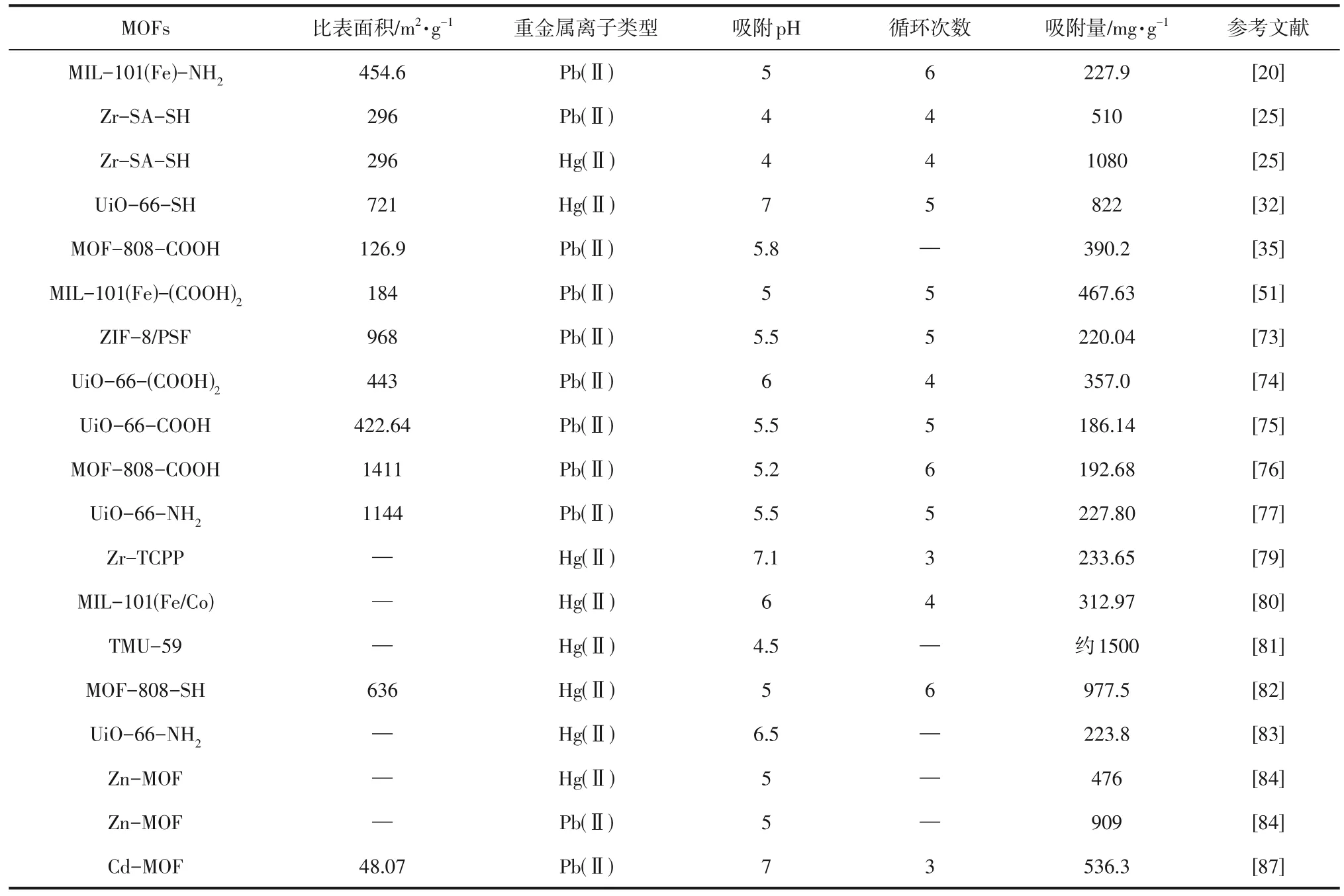
表2 部分MOFs对典型阳离子型重金属的吸附性能
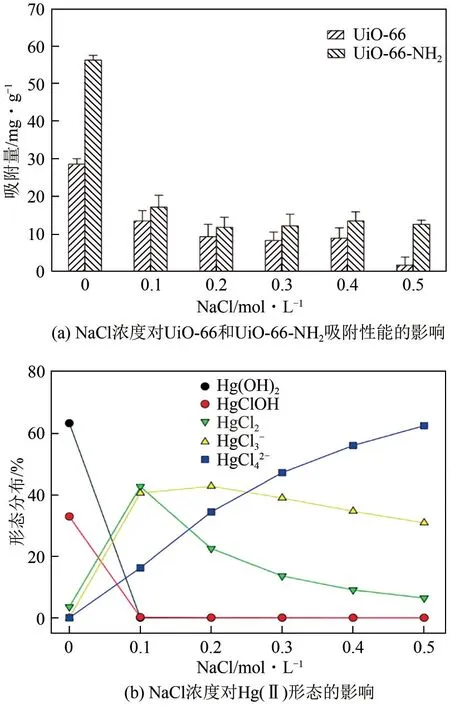
图6 NaCl浓度对Hg(Ⅱ)形态及MOFs吸附性能的影响[82]
3 MOFs 对阴离子型含氧离子的吸附性能
3.1 铬(Cr)
Cr 是动植物的必要微量营养元素之一,相对原子质量为52.01,但是过量的Cr 具有较强的生物毒性,其中Cr(Ⅵ)的毒性约是Cr(Ⅲ)的100倍。Cr(Ⅵ)在水体中的分布形态与pH、浓度有关,Noraee等[85]发现UiO-66 在弱酸性条件下对低浓度HCrO4-具有更强的吸附能力,此时UiO-66 显较强的正电性。由于UiO-66具有较为致密的孔隙,当pH约为2 时,HCrO4-转化为H2CrO4,此时主要发生H2CrO4在MOFs 表面的物理吸附。为了提升在低pH 时对Cr(Ⅵ)的吸附性能,Rego 等[86]构筑了一种Ce(Ⅲ)掺杂的UiO-66,配位数的不同使得UiO-66微孔结构中出现一定量的介孔,较大孔径介孔结构的生成使得H2CrO4在酸性条件仍可以沉积在Ce-UiO-66 的孔隙中,表现出了对Cr(Ⅵ)极强的吸附能力。但是在碱性条件下,UiO-66 稳定性较差,因此可使用ZIF-8 对Cr(Ⅵ)进行吸附,此时Cr(Ⅵ)的形态为CrO42-[85]。
在pH 小于2 时,或Cr(Ⅵ)浓度超过1.0mol/L时,Cr(Ⅵ)的形态转化为体积更大的Cr2O72-,为了促进Cr2O72-在MOFs 中的扩散与结合,需要构筑孔隙更大的MOFs。Zheng等[87]使用均苯三甲酸、4,4′-双咪唑联苯和Cd(Ⅱ)构筑了一种空间群为P21/n的Cd-MOF,该MOFs中每个Cd(Ⅱ)与2个均苯三甲酸的O原子、2个水分子的O原子和2个4,4′-双咪唑联苯的N原子配位,在Cd-MOF中有一个1.60439nm×1.43110nm的三维蛇形空腔,这种特殊的结构使得Cr2O72-可以很好地在MOFs 内部扩散并进行吸附。除此之外,使用氨基化MOFs,如UiO-66-NH2,利用携带正电荷的质子化氨基与Cr(Ⅵ)的静电相互作用也可实现在较低的pH 下吸附Cr(Ⅵ)[图7(a)],该MOFs 循环使用5 次后仍可保持77.0%的吸附效率[88]。但 是UiO-66-NH2的 稳 定 性 较Cd-MOF 更差[87-88]。Li等[89]在UiO-66结构的基础上,外延构筑了UiO-67,这种UiO-66@UiO-67 的核壳结构被证实具有更好的水稳定性与刚性,并可在更广泛的pH范围内高效吸附超过120mg/g的Cr(Ⅵ) [图7(b)]。较好的水稳定性和刚性赋予了UiO-66@UiO-67 较好循环使用性,5 次吸附-解吸循环后仍可恢复90.14%的活性吸附点位。此外,还可将Cr(Ⅵ)还原为Cr(Ⅲ),而后通过静电相互作用-配位作用将其固定在MOFs上[24]。
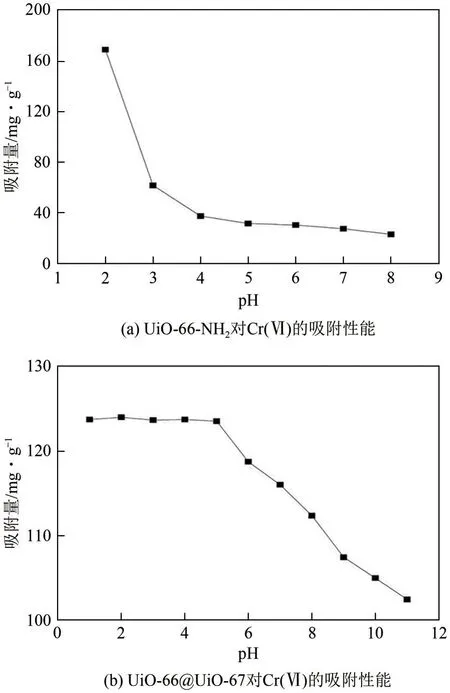
图7 MOFs对Cr(Ⅵ)的吸附性能[88-89]
溶液中的盐可能会干扰MOFs 与Cr(Ⅵ)之间的吸引力,溶液中的阴离子HPO42-和H2PO4-对MOFs吸附性能影响最大,CO32-、SO42-等二价阴离子次之,NO3-和Cl-对MOFs的影响最低[87-90]。总的来说,pH 和盐浓度在改变吸附剂表面电荷、吸引或排斥Cr(Ⅵ)方面起着重要作用,Cr(Ⅵ)在MOFs表面的相互作用取决于pH 和盐浓度的变化,根据电荷变化存在着阴离子-π 电子云、静电吸引、静电排斥和物理填充等相互作用[91]。
3.2 砷(As)
As 属于类重金属,具有两性元素的性质,相对原子质量为74.92,As 会抑制酶的生物活性造成代谢阻碍。As在水体中的存在形式主要是As(Ⅲ)和As(Ⅴ)。As(Ⅲ)的形态与pH有关,As(Ⅲ)在pH小于9.1 时主要以H3AsO3形态存在。Pervez 等[92]发现,在弱碱性条件下H3AsO3更易与MOF-808 发生配体交换,其吸附性能随表面羧基含量的提升而增强;在pH大于9.1时,As(Ⅲ)则以H3AsO2-形式存在,可以通过静电相互作用和阴离子-π 电子云间的相互作用吸附在MOFs 上,但是对MOFs 的耐碱能力有较高的要求。MOFs 中还会存在氧化性较强的官能团或金属离子,例如含有Mn(Ⅳ)的MOFs 在酸性条件下可将还原性较强的As(Ⅲ)氧化为As(Ⅴ),但是这种氧化还原反应会造成MOFs 结构的不可逆缺陷[93-94]。此外,在较低的pH下,Mn(Ⅳ)还原为水溶性的Mn(Ⅱ)会占据吸附剂上的吸附位点,阻碍了Mn(Ⅳ)与As(Ⅲ)之间的电子转移[94]。
As(Ⅴ)的形态与溶液的pH 有关,分别为H3AsO4(pH<1.89)、 H2AsO4-(1.89<pH<4.16)、HAsO42-(4.16<pH<9.20)和AsO43-(pH>9.20)[92]。氨基化改性可以提升MOFs的正电性从而提升对As(Ⅴ)的吸附性能,Yin 等[95]发现,MIL-101(Fe/Al)-NH2可在pH为3~11的范围内高效吸附超过90%的As(Ⅴ),酸性条件下主要依赖的是质子化的氨基与As(Ⅴ)的静电相互作用。随着pH 的增加,MIL-101(Fe/Al)-NH2仍可保持对As(Ⅴ)的高效吸附,这是因为较高的pH下,As(Ⅴ)还可通过Fe(Ⅲ)表面的羟基配体交换被吸附,—FeOH 与As(Ⅴ)的反应较为复杂,根据pH 的变化呈现非、单质子化形式的双齿内层吸附,可生成—FeH2AsO4、—FeHAsO4-、—FeAsO42-和—FeOHAsO43-等稳定的Fe-As 内-球体表面配位化合物[93-96]。在另一个高效吸附As(Ⅲ)/As(Ⅴ)的UiO-66(Fe/Zr)中,发现该MOF 具有较好的循环使用性能,吸附-解吸5 次循环后仍可保持超过90%的吸附性能[96]。除了氨基化改性外,在最新的研究中也发现了羧基化MOFs 对As(Ⅴ)的高效吸附,虽然羧基与As(Ⅴ)均显负电性,但是羧基仍可以通过氢键作用吸附As(Ⅴ)[97]。若同时对MOFs 羧基化、氨基化修饰,则羧基、氨基还可形成稳定的氢键网络并协同吸附As(Ⅴ),氢键网络有效地削弱了有机配体的构象扭曲,增强了MOFs的刚性,并提升了MOFs的水稳定性[98]。
水体中的含氧酸根离子对MOFs 吸附As(Ⅲ)/As(Ⅴ)会造成一定的影响,其中HCO3-、CO32-会与As(Ⅲ)/As(Ⅴ)形成竞争吸附,这是因为相较于As(Ⅲ)/As(Ⅴ)而言,HCO3-、CO32-与金属离子、质子化氨基反应速度更快并可形成稳定的产物;SO42-、NO3-对吸附As(Ⅲ)/As(Ⅴ)的影响较低,这可能是因为SO42-、NO3-与MOFs 的相互作用仅存在氢键和静电吸引作用;HPO42-和PO43-则是会显著影响吸附性能,且随着HPO42-和PO43-浓度的升高吸附量迅速下降,这是因为HPO42-和PO43-具有与As(Ⅲ)/As(Ⅴ)相似的结构和性质,且反应活性更强,HPO42-和PO43-甚至可以置换吸附在MOFs 表面的As(Ⅲ)/As(Ⅴ)[15,93,95-96]。除此之外,SiO32-形成的多硅酸盐层会包覆MOFs的活性吸附位点,钝化MOFs 并造成吸附性能的下降,如图8 所示[96]。部分MOFs 对典型的阴离子型含氧离子Cr(Ⅵ)和As(Ⅲ)/As(Ⅴ)的吸附性能见表3。
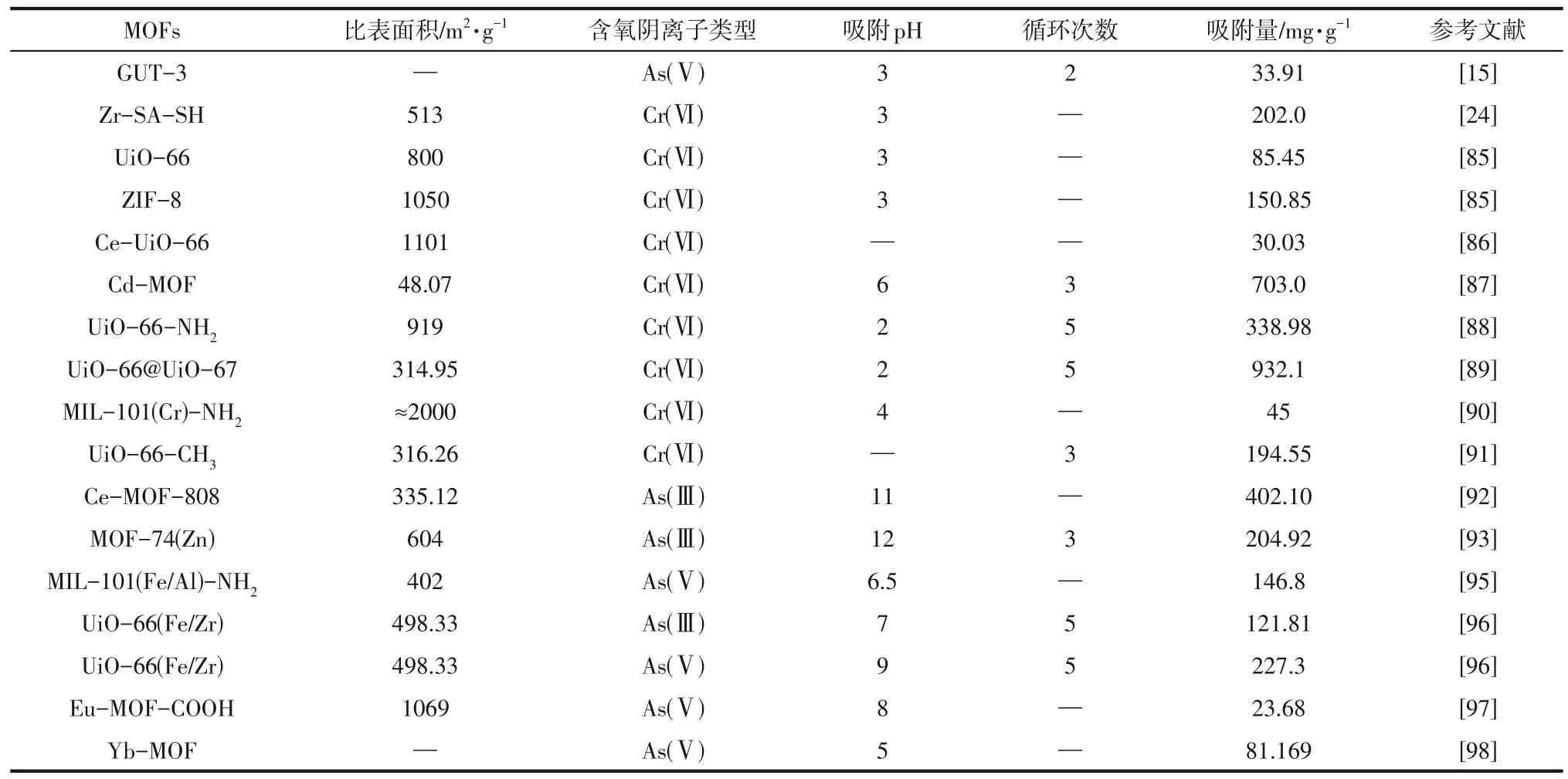
表3 部分MOFs对典型阴离子型含氧离子的吸附性能
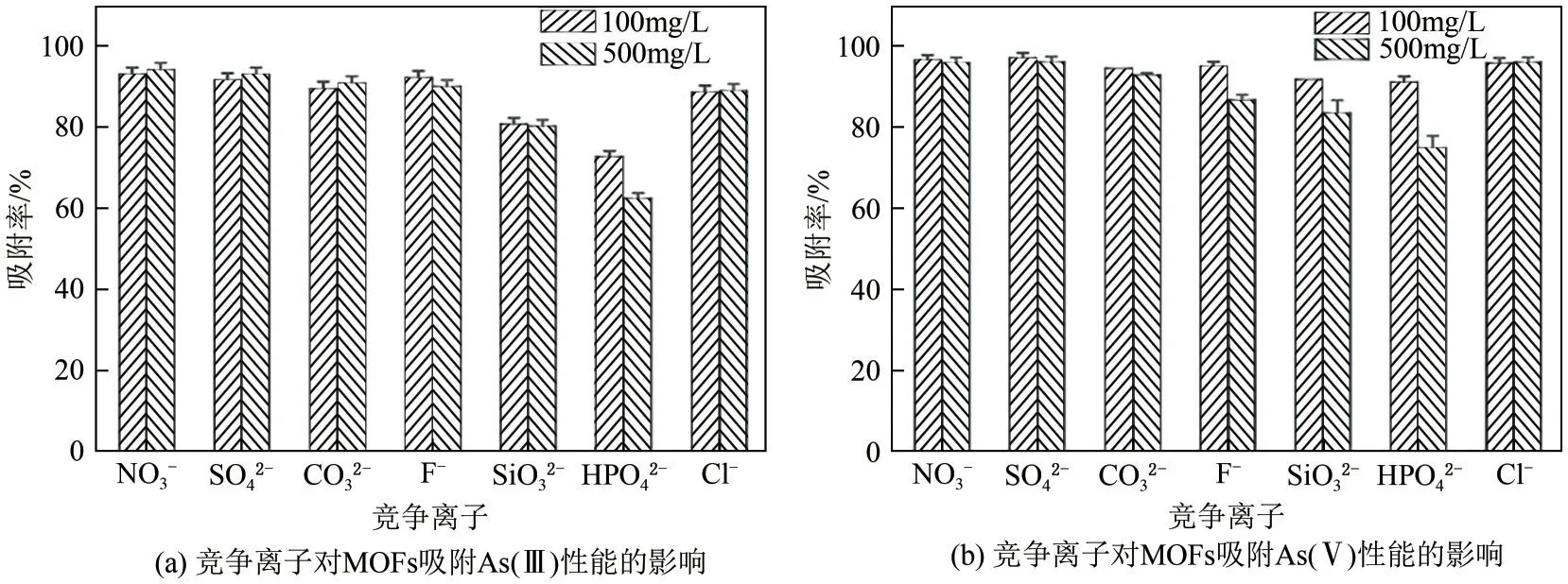
图8 竞争离子对MOFs吸附As性能的影响[96]
4 结语
MOFs 具有的较高比表面积、合适的孔隙结构、较强的稳定性等物理化学特性,加之MOFs上均匀分布的活性官能团、活泼的金属节点,使其具有易于构筑功能化MOFs的优势,在吸附分离水体中的重金属离子方面表现出了巨大的潜力。在MOFs 的众多特性中,多孔性、表面活性和框架柔性是MOFs作为吸附剂的核心性能,水稳定性、可扩展性、生物毒性和循环使用性则是MOFs作为高性能水处理领域的关键。通过对金属离子、有机配体及合成方法的调控,可以实现MOFs在某几个性能上的突破。尽管近年来对MOFs的制备方法、性能研究和多维度应用均取得了一定进展,但将MOFs 实际应用于吸附去除重金属离子仍面临着许多需要解决的问题。
(1)尽管MOFs 在中性、酸性、碱性溶液中表现出了一定的稳定性,但是在实际水体中,含有配位性更强的有机物和金属离子,这种配体或金属离子交换会破坏MOFs 的结构。同时,为了提高MOFs 的吸附量,通常会改变合成条件或功能化修饰使其暴露更多的活性官能团,但是这种具有较高吸附量的MOFs常常具有较明显的结构缺陷,而有结构缺陷的MOFs往往意味着水稳定性和循环使用性的损失,因此仍需深入研究MOFs的吸附性能与水稳定性的平衡关系。
(2)尽管MOFs 具备优异的多孔性、表面活性、框架柔性、水稳定性、可扩展性、循环使用性和较低的生物毒性,但是应用于水处理的MOFs几乎难以同时拥有以上的所有性能,例如可扩展性与表面活性、水稳定性与表面活性、框架柔性与水稳定性、框架柔性与循环使用性、生物毒性与多孔性等常常难以得兼,如何平衡MOFs各性能之间的关系,制备高性能水处理用吸附剂,仍需继续深入研究。
(3)目前应用于重金属吸附的MOFs 多为微小的粉状颗粒,对重金属离子的吸附仍处于实验室探索阶段,对MOFs的生物毒性和可扩展性研究仍不成熟,因此仍需分析其在生态环境中的迁移、循环、富集等环境效应机制,以及大面积的MOFs低成本高效益的可控性制备。
猜你喜欢
机械工业标准化与质量(2022年9期)2022-09-30
石油沥青(2021年5期)2021-12-02
文化创新比较研究(2020年7期)2021-01-13
材料科学与工程学报(2016年4期)2017-01-15
中国卫生(2016年7期)2016-11-13
合成化学(2015年4期)2016-01-17
中国卫生(2015年10期)2015-11-10
中国水利(2015年4期)2015-02-28
科学中国人(2015年22期)2015-02-28
无机化学学报(2014年6期)2014-02-28
