
QuEChERS-液相色谱-串联质谱法测定蜂产品中克百威和3-羟基克百威的残留量
2024-01-08 23:21黎天勇吴舒悦
理化检验-化学分册 2023年12期
黎天勇,李 菊,吴舒悦,席 静,王 岚
(广州海关技术中心,广州 510623)
农药对动物源食品的污染主要来源于饲料中农药的残留,饲料中的农药污染主要源自饲料加工原料(大豆、玉米等)生长过程的农药施用[1-2]。据有关研究显示,氨基甲酸酯类农药如克百威及其代谢物为代表的农药作为有机氯农药的替代品在种植业上已经得到广泛应用[3-4]。克百威又名呋喃丹,是一种具有杀虫和杀螨作用的农药。克百威及其代谢物对病虫胆碱酯酶具有不可逆地抑制作用[5],对多种病虫害防治作用显著。目前,克百威及其代谢物在防治农作物病虫害,控制人畜传染病,提高农畜禽产品产量与质量方面起到举足轻重的作用。但是,由于克百威及其代谢物不可逆地抑制胆碱酯酶,在玉米、小麦等饲料作物中的克百威及其代谢物的残留物可通过食物链进入禽畜及人体内富集累积,从而对禽畜及人体健康、生态环境造成危害[6-8]。近年来,氨基甲酯类农药克百威的过量使用造成中毒的现象时有发生[9-10],因此各国都制定了相关的限量标准。我国GB 2763-2021«食品安全国家标准 食品中农药最大残留限量标准»规定,克百威主要用途为杀虫剂,克百威残留量按“克百威及3-羟基克百威之和,以克百威表示”的规定,而农业部235号公告附录4则将克百威列为禁止在动物中使用的药物种类;欧盟(EU)2015/399 规定,蜂及蜂产品中克百威限量为0.05 mg•kg-1,为临时限量。
当前,氨基甲酸酯类杀虫剂的检测方法涉及比较多的基质,以植物源性基质[11-20]研究为主,如果蔬、中药等,还有土壤[21]、水[22-24]、饮料[25]等基质,动物源性基质的相关研究相对较少[26-27]。现行检测动物性食品中克百威的方法主要有 GB 31658.10-2021«食品安全国家标准 动物性食品中氨基甲酸酯类杀虫剂残留量的测定 液相色谱-串联质谱法»、GB/T 5009.163-2003«动物性食品中氨基甲酸酯类农药多组分残留高效液相色谱测定»、SN/T 2560-2010«进出口食品中氨基甲酸酯类农药残留量的测定 液相色谱-质谱/质谱法»,这3 个标准所适用范围均未涉及蜂产品。蜂产品中氨基甲酸酯类杀虫剂克百威的相关分析研究较少[28-29],且这些方法均不涉及克百威的代谢产物3-羟基克百威,因此适用范围并不广泛。国家标准GB 23200.99-2016«蜂王浆中多种氨基甲酸酯类农药残留量的测定 液相色谱-质谱/质谱法»也只测定克百威原药,没有测定代谢物3-羟基克百威。另外,前处理方法多是采用固相萃取法,在净化过程中提取液经过固相萃取柱净化,耗费溶剂量相对较大,耗时相对较长[26-29]。本工作采用液相色谱-串联质谱法,结合QuECh ERS 前处理技术测定蜂产品中克百威和3-羟基克百威的残留量,可为蜂产品中克百威和3-羟基克百威残留量的检测提供方法参考。
1 试验部分
1.1 仪器与试剂
TSQ QUANTIVA 型液相色谱-质谱/质谱仪;BT223S型电子天平(精确到0.01 g);CPA225D 型电子天平(精确到0.01 mg);ST16R 型台式冷冻离心机;ET3301 型全自动氮吹浓缩仪;Promax 2020型水平往复式振荡器,Multi reax型涡旋振荡器;JYS-M01型磨粉机;Milli-Q 型纯水系统。
克百威标准储备溶液:1 000 mg•L-1,准确称取10 mg的克百威标准物质,用乙腈溶解并定容至10 mL,配制成质量浓度为1 000 mg•L-1的克百威标准储备溶液。
3-羟基克百威标准储备溶液:1 000 mg•L-1,准确称取10 mg的3-羟基克百威标准物质,用乙腈溶解并定容至10 mL,配制成质量浓度为1 000 mg•L-1的3-羟基克百威标准储备溶液。
克百威、3-羟基克百威标准物质的纯度不小于98%;乙腈为色谱纯;无水硫酸镁、氯化钠、氢氧化钙均为分析纯;乙二胺-N-丙基硅烷化硅胶(PSA)粒径为40~60μm;十八烷基硅烷键合硅胶(C18)粒径为50μm;试验用水为超纯水。
1.2 仪器工作条件
1.2.1 色谱条件
Accucore aQ C18色谱柱(150 mm×2.1 mm,2.6μm);柱温45 ℃;流量0.3 mL•min-1;进样量2μL;流动相A 为含5 mmol•L-1甲酸铵的0.1%(体积分数,下同)甲酸溶液;B为甲醇。梯度洗脱程序:0~5.0 min 时,B 由20% 升至90%,保持3.0 min;8.0~9.0 min时,B由90%降至20%,保持1.0 min。
1.2.2 质谱条件
电喷雾电离(ESI)源,正离子(ESI+)扫描;鞘气压力241.32 kPa,辅助气压力68.95 kPa,吹扫气压力6.89 kPa;离子传输管温度350 ℃,蒸发温度300 ℃;多反应监测(MRM)方式。其他质谱参数见表1,其中,“∗”代表定量离子。

表1 质谱参数Tab.1 MS parameters
1.3 试验方法
1.3.1 提取
称取蜂蜜、蜂王浆2 g,分别置于50 mL离心管中,加5 mL水,涡旋混匀1 min,静置5 min。加入10 mL乙腈,涡旋混匀1 min,摇床振荡10 min,加入2~3 g 氯化钠之后,涡旋均匀1 min,以转速4 000 r•min-1离心5 min,分别取出1 mL 蜂蜜、蜂王浆上清液作为待净化液。
称取蜂胶0.2 g 于50 mL 离心管中,加入10 mL乙腈,涡旋混匀1 min,置超声仪提取10 min,加入10 mL 0.5%(质量分数)氢氧化钙溶液,涡旋混匀2 min后再加4~6 g氯化钠,涡旋混匀2 min,于0 ℃以转速10 000 r•min-1离心10 min,取4 mL蜂胶上清液作为待净化液。
1.3.2 净化
取上述蜂蜜待净化液、蜂王浆待净化液,分别转移至内含150 mg无水硫酸镁、50 mg C18和50 mg PSA 的高速离心管中,涡旋均匀约1 min,以转速12 000 r•min-1离心5 min。分别取蜂蜜、蜂王浆的上清液过0.22μm 有机相滤膜后按照仪器工作条件测定。
取蜂胶待净化液全部转移至内含除水剂和净化材料的塑料离心管中(每毫升待净化液使用150 mg无水硫酸镁、50 mg C18和50 mg PSA),涡旋均匀后,以转速12 000 r•min-1离心5 min。定量吸取2 mL上清液至氮吹管,于45 ℃水浴氮吹至干后立即加入1 mL乙腈复溶,涡旋混匀,过0.22μm 有机相滤膜后按照仪器工作条件进行测定。
2 结果与讨论
2.1 前处理条件的选择
2.1.1 提取溶剂
在氨基甲酸酯类药物残留检测研究中,乙腈、丙酮、乙酸乙酯是比较常用的提取溶剂。试验以蜂蜜、蜂王浆和蜂胶样品为研究对象,蜂蜜、蜂王浆中克百威和3-羟基克百威的加标量均为0.01 mg•kg-1,蜂胶中克百威和3-羟基克百威的加标量均为0.05 mg•kg-1,分别各用10 mL乙腈、含1%(体积分数,下同)乙酸的乙腈溶液、乙酸乙酯、丙酮作为提取溶剂,考察了提取溶剂对蜂蜜、蜂王浆和蜂胶样品克百威和3-羟基克百威回收率的影响,结果见图1和图2。

图1 提取溶剂对克百威回收率的影响Fig.1 Effect of extraction solvents on recovery of carbofuran
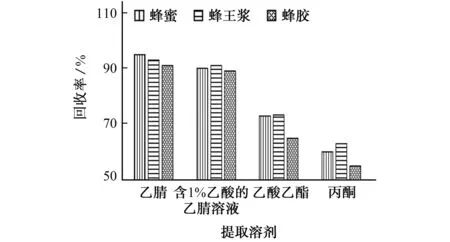
图2 提取溶剂对3-羟基克百威回收率的影响Fig.2 Effect of extraction solvents on recovery of 3-hydroxycarbofuran
由图1、图2可知:以乙腈和含1%乙酸的乙腈溶液为提取溶剂时,克百威和3-羟基克百威的回收率均能达到90.0%以上;以乙酸乙酯和丙酮为提取溶剂时,克百威和3-羟基克百威的回收率均偏低,而且提取液整体比较浑浊,判断是提取过程中被溶解出来的杂质偏多,这给后续净化带来困难。
以蜂胶为研究对象,在使用酸性乙腈为提取溶剂时,分离度不好,提取液整体浑浊;而使用乙腈提取时,提取液颜色较浅,杂质相对较少,分层效果明显。因此,试验选择乙腈作为提取溶剂。
2.1.2 净化剂
常用的净化方式包括Qu ECh ERS净化和固相萃取净化,考虑到固相萃取柱需要活化和洗脱程序,过程繁琐,溶剂耗费量大,因此采用更能节约溶剂,操作更为简便的 Qu ECh ERS 净化方式。Qu ECh ERS净化方式中常用的净化剂有PSA、C18、石墨化碳黑(GCB)等。PSA 和C18在化学分析中被广泛使用,是去除基质中的脂肪、糖类和有机酸等的良好材料;GCB 由于其特殊结构,使得其对色素和某些平面对称结构物质有吸附作用。
试验选用3种净化方案,方案一(150 mg无水硫酸镁+50 mg PSA+50 mg C18+25 mg GCB)、方案二(150 mg无水硫酸镁+50 mg PSA+50 mg C18)、方案三(150 mg无水硫酸镁+50 mg PSA)。考察了上述3种净化方案对蜂蜜中克百威和3-羟基克百威回收率的影响,结果见图3。

图3 净化方案对克百威和3-羟基克百威回收率的影响Fig.3 Effect of purification methods on recovery of carbofuran and 3-hydroxycarbofuran
由图3 可知,Qu ECh ERS 方案二的回收率较高,达到90.0%以上。因此,试验选择的净化剂为150 mg无水硫酸镁+50 mg PSA+50 mg C18。
2.2 色谱条件的选择
试验比较了甲醇-0.1% 甲酸溶液、甲醇-含5 mmol•L-1甲酸铵的0.1%甲酸溶液等流动相体系的分离效果,结果表明,克百威与3-羟基克百威在甲醇-含5 mmol•L-1甲酸铵的0.1%甲酸溶液流动相体系中离子化程度更高、响应值更大,峰形更好。综合考虑,试验选择甲醇-含5 mmol•L-1甲酸铵的0.1%甲酸溶液体系为流动相。
试验考察了柱温(30,35,40,45 ℃)对分离效果的影响,结果发现,柱温对克百威和3-羟基克百威的色谱分离效果影响不大,但柱温越高,色谱分离的速率越快。因此,为节省分析时间,试验选择45 ℃作为柱温。
在优化的条件下,克百威出峰时间为5.10 min,3-羟基克百威出峰时间为3.81 min。0.01 mg•L-1克百威标准溶液和0.01 mg•L-13-羟基克百威标准溶液的MRM 色谱图见图4。

图4 色谱图Fig.4 Chromatograms
2.3 质谱条件的选择
以10μL•min-1的流量将1.0 mg•L-1的克百威和3-羟基克百威标准溶液分别连续注入ESI源中,在ESI+模式下对克百威和3-羟基克百威进行一级质谱分析(Q1扫描),得到克百威和3-羟基克百威的准分子离子,并优化去簇电压等参数。对准分子离子进一步采用二级质谱分析(子离子扫描),得到相关子离子信息,通过进一步优化碰撞能量,克百威与3-羟基克百威的二级质谱见图5。选择信号最强、无干扰的离子对作为定量离子对,选响应值高,m/z大且基线噪声相对低的离子对作为定性离子对。此外,对离子源、鞘气、辅助气等参数进行优化,采用MRM 模式采集数据,优化的质谱条件详见1.2.2节。

图5 二级质谱图Fig.5 Secondary mass spectra
2.4 基质效应
基质效应(ME)指的是基质对分析过程的干扰及对分析结果的影响,其计算公式为ME=(A1-A2)/A2×100%,其中A1、A2 分别为各目标化合物在基质与纯溶剂中的响应值(即峰面积)。|ME|大于50%通常被认为基质效应明显;|ME|在20%~50%内则被认为基质效应中等;|ME|小于20%被认为存在弱基质效应。试验中,按照仪器工作条件分别测定纯溶剂乙腈配制的克百威和3-羟基克百威标准溶液、基质(蜂蜜、蜂王浆、蜂胶)匹配的克百威和3-羟基克百威标准溶液,记录目标物的峰面积,计算ME,结果见表2。

表2 基质效应Tab.2 Matrix effect
由表2可知,3种基质下均存在弱基质效应,对两种目标物测定影响不大。考虑到蜂蜜产品基质复杂,采用基质匹配的标准溶液系列建立工作曲线。
2.5 工作曲线和测定下限
分别以空白蜂蜜、蜂王浆、蜂胶为基质,按照试验方法处理,加入克百威和3-羟基克百威标准储备溶液,用乙腈逐级稀释,配制成质量浓度分别为0.002,0.005,0.010,0.020,0.050,0.100 mg•L-1的基质匹配的克百威和3-羟基克百威标准溶液系列。以目标物的质量浓度为横坐标,对应的峰面积为纵坐标绘制工作曲线。结果表明,克百威和3-羟基克百威的质量浓度均在0.002~0.100 mg•L-1内与对应的峰面积呈线性关系,所得线性回归方程和相关系数见表3。

表3 线性参数和测定下限Tab.3 Linearity parameters and lower limits of determination
对于试验中测定下限的设置,首先考虑的是对应浓度水平信噪比(S/N),回收率和精密度是否符合相关要求;其次考虑的是各个国家最严格的限量。最终通过在空白基质中添加目标物组分,以10倍信噪比对应的浓度水平得到测定下限(10S/N),结果见表3。
2.6 精密度和回收试验
在蜂蜜、蜂王浆和蜂胶空白样品中定量添加克百威和3-羟基克百威标准溶液,按照试验方法对每个浓度水平重复测定6次,计算回收率和测定值的相对标准偏差(RSD),结果见表4。

表4 精密度和回收试验结果(n=6)Tab.4 Results of tests for precision and recovery(n=6)
由表4可知,克百威的回收率为88.8%~108%,测定值的RSD 为0.50%~3.5%;3-羟基克百威的回收率为84.5%~106%,测定值的RSD 为2.0%~6.8%。说明方法的精密度和准确度良好。
2.7 样品分析
随机抽取市售蜂产品共6份,按照试验方法进行分析,结果显示,所抽取样品中,均未检出克百威和3-羟基克百威。
为解决现行动物源性食品中检测克百威和其代谢物3-羟基克百威标准中缺少蜂产品的问题,本工作采用液相色谱-串联质谱法,结合QuEChERS前处理技术测定蜂产品中克百威和3-羟基克百威的残留量。采用的QuECh ERS方法一定程度上简化了样品处理流程,成本更低,耗费溶剂量相对较少,环境友好;同时,本方法的测定下限能满足我国农业部235号公告及欧盟(EU)2015/399对蜂产品的相关规定要求。
猜你喜欢
科学与财富(2021年34期)2021-05-10
浙江农业学报(2020年11期)2020-12-02
中成药(2018年2期)2018-05-09
益寿宝典(2017年35期)2017-08-21
新乡学院学报(2016年6期)2016-12-01
当代化工研究(2016年9期)2016-03-20
食品科学(2013年6期)2013-03-11
河北大学学报(自然科学版)(2012年3期)2012-03-25
中国蜂业(2010年1期)2010-08-15
祝您健康(1999年4期)1999-12-25
