
自动固相萃取-超高效液相色谱-串联质谱法同时测定暂养水中45种抗生素药物的含量
2024-01-08 23:21刘奕雄张凯旋朱雨田冯玉桢袁定帅赵博扬杜业刚
理化检验-化学分册 2023年12期
刘奕雄,张凯旋,朱雨田,李 意,冯玉桢,袁定帅,赵博扬,杜业刚
(深圳市计量质量检测研究院,深圳 518109)
氯霉素、孔雀石绿等药物都是人工合成抗生素[1-2],常被用作治理水产养殖中真菌、细菌及寄生虫感染的首选药物。硝基呋喃类、氯霉素类等抗生素早在2002年就均被我国农业部门列入«食品动物禁用的兽药及其它化合物清单»,但是每年水产品抽检中还是能发现违规使用禁用药物问题[3],长期食用不合格水产品会危害人体健康[4-5]。为了降低水产品发病率、提高存活率,不法商贩可能会向暂养水中违规投放抗生素药物,这也是导致水产品中检出违禁品和抗生素药物残留超标的原因之一。
目前检测抗生素的方法主要是酶联免疫法[6-8]、毛细管电泳法[9-11]、液相色谱法[12]、液相色谱-串联质谱法[13-18]等。不同抗生素药物化学结构和性质差异较大,存在同时处理检测复杂且耗时长的问题,导致一次检测药物种类少,回收率不理想。本工作针对目前水产品检出率较高的喹诺酮类、孔雀石绿、四环素类、磺胺类、呋喃类、氯霉素类等6类抗生素展开方法学研究,提出了自动固相萃取-超高效液相色谱-串联质谱法(SPE-UHPLC-MS/MS)测定水产品暂养水中45种抗生素药物含量的方法,具有分析效率高、重复性高、检测时间短的特点,实现了水产品暂养水的高通量抗生素检测。
1 试验部分
1.1 仪器与试剂
Waters Acquity UPLCⓇI-Class型超高效液相色谱仪;AB SCIEX Triple QuadTM5500型质谱仪;Fotector Plus型全自动高通量固相萃取仪;HLB固相萃取柱(60 mg/3 mL);Auto EVA-60 型控温氮吹浓缩仪;QUINTIX224-1CN 型电子天平;Milli-Q Direct 8型超纯水系统。
单标准储备溶液:100 mg•L-1,称取适量的各标准物质(相当于活性成分10 mg),用甲醇溶解,并定容至100 mL,配制成质量浓度为100 mg•L-1的单标准储备溶液。
混合内标溶液:取适量的内标的单标准储备溶液,用20%(体积分数,下同)甲醇溶液稀释,配制成氘代喹诺酮类、氘代磺胺类的质量浓度为1.00 mg•L-1,氘代孔雀石绿、氘代隐色孔雀石绿、氘代氯霉素类的质量浓度为100μg•L-1的混合内标溶液。
乙二胺四乙酸二钠(Na2EDTA)-Mcllvaine缓冲溶液:将1 L 的0.1 mol•L-1柠檬酸溶液与625 mL的0.2 mol•L-1磷酸氢二钠溶液混合,调节pH 为(4.0±0.5);称取60.5 g Na2EDTA 放入上述1 625 mL 缓冲溶液中,使其溶解混匀,配制成0.1 mol•L-1Na2EDTA-Mcllvaine缓冲溶液。
4种四环素类、3种氯霉素类、15种喹诺酮类、17种磺胺类、4种呋喃类、孔雀石绿及代谢物隐色孔雀石绿、8种氘代化合物的标准物质的纯度均大于98%;甲醇、甲酸为色谱纯;Na2EDTA 为分析纯;试验用水为一级水。
1.2 仪器工作条件
1.2.1 色谱条件
Agilent Eclipse Plus C18RRHD 色谱柱(100 mm×2.1 mm,1.8μm);柱温35 ℃;进样量4μL;流量0.3 mL•min-1;流动相A 为0.1%(体积分数,下同)甲酸溶液,B为甲醇。梯度洗脱程序:0~3.5 min时,A 由95%降至70%;3.5~7.0 min时,A 由70%降至5%,保持1.0 min;8.0~9.0 min时,A 由5%升至95%,保持1.0 min。
1.2.2 质谱条件
电喷雾离子(ESI)源,正离子(ESI+)、负离子(ESI-)分段时间扫描监测;多反应监测(MRM)模式;离子源电压5 500 V/-4 500 V;离子源温度500 ℃;气帘气压力275.8 kPa;雾化气压力344.75 kPa;辅助加热气压力344.75 kPa。53种化合物的保留时间、碰撞能量、去簇电压等参数见表1,其中“∗”代表定量离子对。

表1 质谱参数Tab.1 MS parameters
1.3 试验方法
1.3.1 样品采集
水产品养殖水取自餐饮环节和流通环节,抽取暂养池水500 mL,置于4 ℃冰箱保存。
1.3.2 样品前处理
准确移取40.0 mL经0.45μm 尼龙滤膜过滤后的暂养水,与10 mL Na2EDTA-Mcllvaine缓冲溶液混匀,加入0.05 mL 混合内标溶液,用0.1 mol•L-1盐酸溶液调节溶液酸度至pH 4,然后以1滴•s-1的速率过HLB固相萃取柱(预先用3 mL甲醇、3 mL水活化),待样品完全流出后,以2 mL 水淋洗,减压抽干,用6 mL 甲醇洗脱。收集洗脱液,于40 ℃氮吹浓缩至近干,用1.00 mL 20%甲醇溶液溶解残渣,经0.22μm 滤膜过滤,按照仪器工作条件测定。
1.3.3 数据处理
氯霉素类、喹诺酮类、磺胺类、孔雀石绿及代谢物用内标法定量,四环素类和呋喃类药物用外标法定量。用AB SCIEX 数据处理软件对质量浓度、线性回归方程、回收率进行分析,用Excel软件进行净化方式对比、相对标准偏差(RSD)分析。
2 结果与讨论
2.1 前处理条件的优化
2.1.1 水样酸度
暂养水样酸度会影响药物存在形式,影响最终富集净化效果,因此调节溶液酸度显得尤为重要。四环素类药物属于两性化合物,在碱性和高温环境下不稳定,在中性条件下其羟基以及羰基等官能团与游离的重金属离子形成络合物[19]。国家标准方法[20-21]和文献[22-24]选择Na2EDTA-Mcllvaine缓冲溶液(pH 4)提取,在酸性环境中四环素类药物较为稳定,水中的金属钙镁离子优先与缓冲液中的EDTA 发生螯合反应,从而提高药物富集效果。喹诺酮类药物大部分属于两性化合物,结构形态受酸度影响显著[25]。在强酸性条件下,氟喹诺酮以强阳离子形态存在;当pH 为3~4时,氟喹诺酮主要以两性离子形态存在;当pH 进一步升高时,氟喹诺酮以弱阴离子形态存在。将水样酸度调至pH 为2,4,6,8进行加标回收试验,结果发现当pH 为4时四环素类、喹诺酮类、磺胺类药物的回收率较好,其他药物回收率亦满足要求。因此,试验选择水样酸度pH 为4。
2.1.2 固相萃取柱
暂养水中药物含量比较低,需要富集净化才能进行检测[26]。液液萃取操作繁琐,并且四环素类药物的回收率低。样品先经0.45μm 尼龙滤膜过滤,能除掉水中悬浮大颗粒杂质,防止堵塞固相萃取柱。试验采用MCX 固相萃取柱净化时,四环素类、氯霉素类和大多数硝基呋喃类药物回收率为0,而采用HLB固相萃取柱净化时,45种药物的回收率满足60.0%~120%的要求,如图1 所示。HLB 固相萃取柱的吸附剂是由亲脂性二乙烯苯和亲水性N-乙烯基吡咯烷酮两种单体按一定比例聚合成的大孔有机聚合物。二乙烯苯含有亲脂性的官能团,具有反相吸附剂的特性,而N-乙烯基吡咯烷酮含有亲水性的官能团,具备保留极性化合物的能力。通过调节水相条件改变亲水-亲脂平衡,HLB 固相萃取柱适用非极性、中等极性、极性化合物的净化,能对四环素类、氯霉素类、硝基呋喃类药物等进行较好的吸附净化。因此,试验选择HLB 固相萃取柱进行净化。

图1 采用HLB固相萃取柱与MCX 固相萃取柱时45种药物回收率的对比Fig.1 Comparison of recoveries of 45 drugs using HLB and MCX solid phase extraction columns
2.1.3 洗脱剂
试验对比了甲醇、体积比1∶1的甲醇-乙酸乙酯混合液、体积比1∶1的甲醇-丙酮混合液等3种试剂对45种药物的洗脱效果,结果见图2。
由图2可知:采用体积比1∶1的甲醇-乙酸乙酯混合液洗脱时,磺胺嘧啶和呋喃唑酮的回收率大于120%,金霉素的回收率小于60.0%;采用体积比1∶1的甲醇-丙酮混合液洗脱时,土霉素、强力霉素、培氟沙星的回收率大于120%;而采用甲醇洗脱时,45种药物的回收率均在60.0%~120%内。因此,试验选择甲醇为洗脱剂。
2.2 色谱条件的优化
本工作研究涉及了45种抗生素药物,它们之间化学结构和性质有差异,以Agilent Eclipse Plus C18RRHD 色谱柱分离,采用梯度洗脱才能获得分离度较好的色谱峰。试验比较了甲醇-水、乙腈-水、甲醇-5 mmol•L-1乙酸铵溶液、甲醇-0.1%甲酸溶液等流动相体系的分离效果,结果发现采用0.1%甲酸溶液作为流动相的水相,可形成尖而窄的色谱峰,提高ESI+模式下的化合物质谱信号,同时也不影响ESI-模式下的化合物质谱信号,因此试验最终选择甲醇-0.1%甲酸溶液作为流动相体系。45种药物的MRM 色谱图见图3,其中氯霉素和孔雀石绿的质量浓度为2.00μg•L-1,其他药物的质量浓度为10.0μg•L-1。
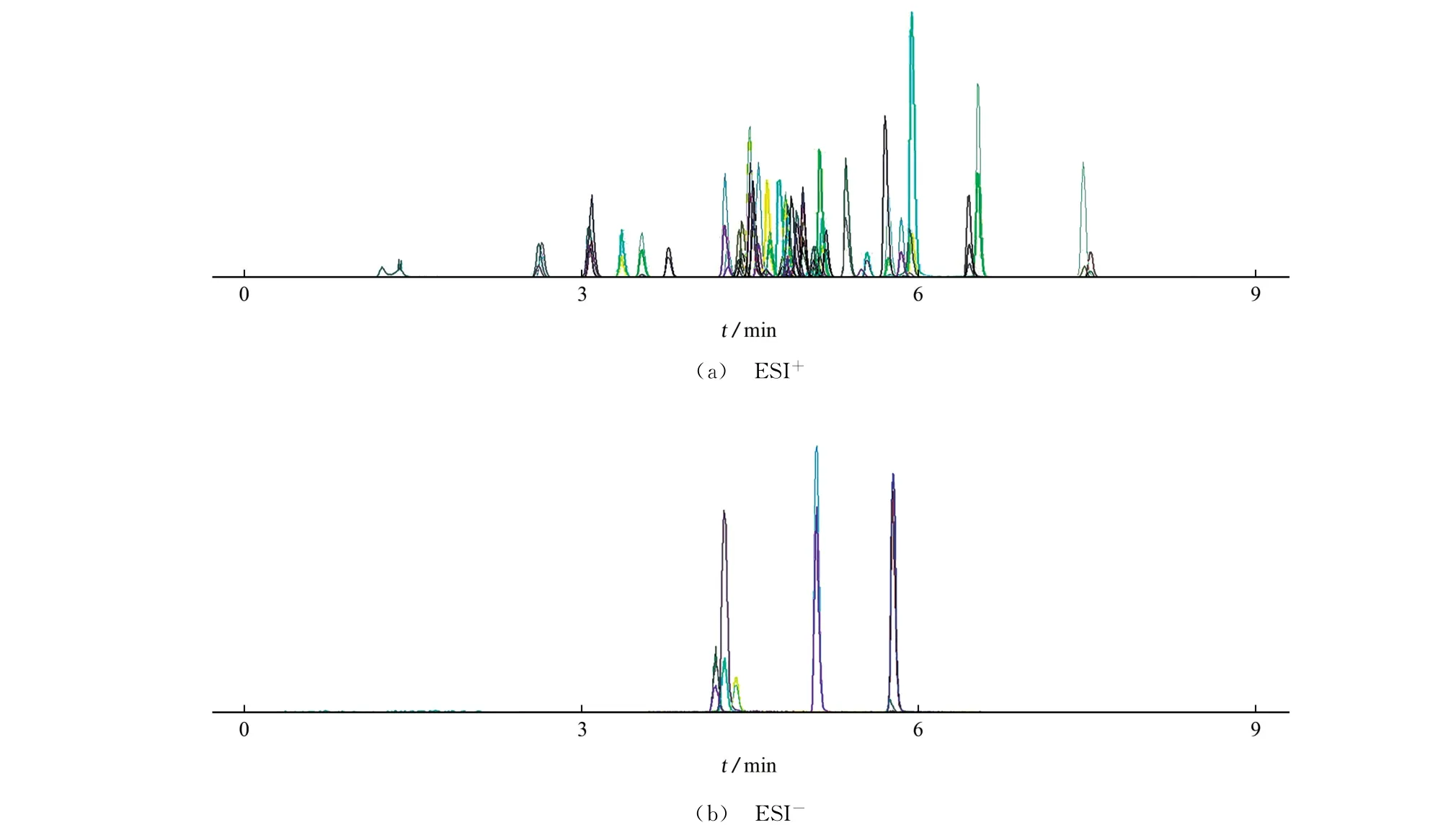
图3 45种药物MRM 色谱图Fig.3 MRM chromatograms of 45 drugs
2.3 质谱条件的优化
分别将0.1 mg•L-1标准溶液在ESI+、ESI-模式下进行母离子全扫描。选择合适电离方式进行质谱分析得到特征碎片离子,进一步优化质谱参数,使特征碎片离子强度达到最大。其中氯霉素、氟甲砜霉素、甲砜霉素、呋喃西林、呋喃妥因在ESI+模式下质谱信号非常弱,反而在ESI-模式下有较强质谱信号,而其他药物正好相反。具体的质谱条件见1.2.2节。
2.4 工作曲线和测定下限
用空白基质提取液逐级稀释,并加入适量混合内标溶液,配制基质匹配的混合标准溶液系列,其中内标氘代喹诺酮类、氘代磺胺类的质量浓度为50.0μg•L-1,氘代孔雀石绿、氘代隐色孔雀石绿、氘代氯霉素的质量浓度为5.00μg•L-1。按照仪器工作条件测定上述溶液,其中四环素类和呋喃类药物以质量浓度为横坐标,峰面积为纵坐标;其他药物以质量浓度为横坐标,以其对应的峰面积与内标峰面积比值为纵坐标,绘制工作曲线。结果显示,氯霉素类和孔雀石绿工作曲线的线性范围为0.200~20.0μg•L-1,其他药物工作曲线的线性范围为1.00~100μg•L-1,相关系数为0.995 1~0.999 9。
参考GB 5009.295-2023«食品安全国家标准化学分析方法验证通则»,向空白样品基质中添加目标分析物,以信噪比为10时的添加浓度水平作为测定下限。结果显示,氯霉素类和孔雀石绿的测定下限为5.0 ng•L-1,其他药物的测定下限为25 ng•L-1。
2.5 精密度和回收试验
取空白水样40.0 mL,置于50 mL 聚丙烯离心管 中,进行低浓度水平0.050 0 μg•L-1或0.250μg•L-1、中浓度水平0.150 μg•L-1或0.750μg•L-1、高浓度水平0.250 μg•L-1或1.25μg•L-1的加标回收试验,每个浓度水平平行测定6 次,计算回收率和测定值的RSD。结果显示:四环素类药物的回收率为68.4%~92.7%,测定值的RSD 为2.5%~4.7%;喹诺酮类药物的回收率为61.6%~106%,测定值的RSD 为1.4%~8.9%;磺胺类药物的回收率为75.7%~112%,测定值的RSD 为1.2%~6.3%;孔雀石绿药物的回收率为85.4%~110%,测定值的RSD 为1.7%~6.0%;呋喃类药物的回收率为80.6%~115%,测定值的RSD 为1.3%~8.4%;氯霉素类药物的回收率为86.8%~115%,测定值的RSD 为1.9%~6.4%。说明方法的精密度和准确度满足分析要求。
2.6 方法比对
本方法与其他文献方法比较结果见表2。结果表明,本方法的测定下限和回收率等能够满足检测水样中多种药物的定量分析要求。相对于传统的手动或者半自动固相萃取装置,自动固相萃取方法进行富集,富集水样体积较少,采用UHPLC 分析时间短,可有效缩短样品测试周期,覆盖药物种类和数量较多,能满足监管部门要求。
2.7 样品分析
覆盖人们日常消费的鲈鱼、基围虾、福寿鱼等31种水产品类别,采集共82批暂养水,采用试验方法对45种药物进行分析,其中21批暂养水中检出药物残留,总检出率为25.6%,详细结果见表3(“∗”代表同批次中同时检测出恩诺沙星和环丙沙星)。其中氯霉素检出量为0.043 0~26.0μg•L-1;恩诺沙星检出量为0.330~200μg•L-1;环丙沙星检出量为0.260~6.40μg•L-1;氟甲砜霉素检出量为2.80μg•L-1;磺胺二甲嘧啶检出量为0.340μg•L-1。从检测结果来看,水产品暂养水中氯霉素、恩诺沙星、环丙沙星检出率较高。流通环节暂养水样品抗生素药物残留检出率为27.5%,占比最高,说明此环节水产品暂养水质量可能存在一定风险。水产品在此环节暂养时间较长,有商家在暂养池水中使用抗生素甚至是违禁药物来抑制相关致病菌繁殖,预防水产品相互摩擦碰撞导致伤口感染病菌,达到保护水产品品质的目的。

表3 暂养水样分析结果Tab.3 Analytical results of temporary aquaculture water samples
本工作采用SPE-UHPLC-MS/MS测定水产品暂养水中45种抗生素药物的含量,具备富集水样体积少、分析简单快速、精密度好等特点,可为相关安全监管部门监测养殖水质量风险提供技术支持。受抽检样品数量所限,今后可适当增加暂养水类别和暂养水批次进行监测分析,以进一步发现暂养水中药物残留水平。
猜你喜欢
食品安全导刊(2021年20期)2021-08-30
中国油脂(2020年3期)2020-04-10
三峡大学学报(自然科学版)(2017年1期)2017-03-20
无机化学学报(2016年8期)2016-12-06
成都信息工程大学学报(2016年6期)2016-06-01
家庭科学·新健康(2016年5期)2016-05-12
化学分析计量(2016年1期)2016-03-14
分析测试学报(2015年3期)2016-01-13
中国药物应用与监测(2015年5期)2015-12-11
中国药业(2014年21期)2014-05-26
