
Ni/CeθM1-θOx催化剂催化二苯并呋喃加氢脱氧性能研究
2024-01-07 07:04韩晶晶杜朕屹
天然气化工—C1化学与化工 2023年6期
韩晶晶,马 柳,杜朕屹
(1.太原理工大学 化学工程与技术学院,山西 太原 030024;2.太原理工大学 省部共建煤基能源清洁高效利用国家重点实验室,山西 太原 030024)
以中低温热解为核心的分质利用是低阶煤清洁高效利用的重要研究方向。中低温煤焦油中杂原子氧的含量较高,会造成油品热值低和稳定性差的问题[1-2]。因此,有必要对中低温煤焦油进行加氢脱氧(HDO)处理,以满足其作为燃料的应用要求。中低温煤焦油中含氧化合物主要以酚类和呋喃类化合物为主[3],其中,二苯并呋喃(DBF)分子量大,反应活性低,氧原子难以脱除[4]。目前,HDO是脱除DBF的氧原子最有效的方法。
WU 等[5]研究了双金属催化剂Pt1Ni4/MgO 对DBF 的HDO 反应的影响,发现Pt 位点具有较强的活化H2的能力,可以实现对DBF的预加氢,Ni位点促进C—O 键的断裂,提升了目标产物环己烷(BCH)收率。WANG 等[6]研究了DBF 在Pt/介孔ZSM-5催化剂上的加氢脱氧反应,发现与Pt/ZSM-5和Pt/Al2O3相比,Pt/介孔ZSM-5呈现出更强的活性,归因于介孔ZSM-5 的强酸性和介孔孔道的协同作用。催化剂的酸性、孔径分布和金属分散度等因素影响了催化剂加氢脱氧反应的性能。目前,研究发现可还原性金属氧化物因其表面原子配位数的差异,易形成表面氧空位,而催化剂表面氧空位对催化反应具有促进作用,引起了广泛关注[7-8]。
XⅠAO等[9]研究了不同晶相的Co/ZrO2对5-羟甲基糠醛加氢脱氧反应的影响,发现C==O 键加氢和C—O键氢解的高活性归因于高浓度的Zr3+-Vo中心与更多酸位点之间的协同作用。REAENDE等[10]制备了Ni/Ce1-xNbxO2催化剂用于苯酚加氢脱氧反应,发现Nb原子插入到CeO2晶格中,促进了Ce4+到Ce3+的还原,形成了催化剂表面缺陷位,有利于反应物的活化。LU等[11]利用共沉淀法制备了Ni/ZrO2-CeO2催化剂,发现ZrO2的存在促进了Ni/ZrO2-CeO2催化剂中氧空位的产生,其氧空位强化了催化剂对愈创木酚中氧原子的吸附与活化,从而提高了催化剂断裂C—O 键的能力。可还原金属氧化物在还原过程中部分还原产生的氧空位可以促进含氧化合物中C—O 键的活化、断裂,从而提高催化剂的催化活性。结合当前催化剂表面氧空位对加氢脱氧反应性能的研究进展,本研究选用DBF 为模型化合物,制备金属离子掺杂的CeθM1-θOx载体并以浸渍法负载Ni 制备Ni/CeθM1-θOx催化剂(M=Ga,Zr,Fe),探究催化剂表面氧空位相对量大小及其对DBF 的HDO反应活性的影响。
1 实验部分
1.1 实验材料与试剂
实验所使用试剂如下:六水合硝酸铈(纯度99.95%,上海阿拉丁生化科技股份有限公司);六水合硝酸镍(纯度99.95%,上海阿拉丁生化科技股份有限公司);九水合硝酸铁(纯度99.95%,上海阿拉丁生化科技股份有限公司);硝酸镓六水合物(纯度99.9%,上海阿拉丁生化科技股份有限公司);硝酸氧锆水合物(分析纯,上海阿拉丁生化科技股份有限公司);氢氧化钠(纯度99.9%,上海阿拉丁生化科技股份有限公司);二苯并呋喃(色谱纯,梯希爱化成工业发展有限公司);正十二烷(色谱纯,上海阿拉丁生化科技股份有限公司);正癸烷(色谱纯,上海麦克林生化科技有限公司);无水乙醇(纯度>99.5%,上海麦克林生化科技有限公司)。
1.2 催化剂制备
1.2.1 载体制备
CeθM1-θOx载体采用水热法制备。θ为CeθM1-θOx载体中Ce的实际物质的量分数,在CeθGa1-θOx、CeθZr1-θOx载体中,θ=0.5;CeθFe1-θOx载体中,θ=0.7。
CeθGa1-θOx载体的制备:称取2.7643 g NaOH 加入10 mL 去离子水中,记为溶液A。分别称取相应质量(n(Ga):n(Ce)=1:1)的硝酸镓六水合物和六水合硝酸铈,并加入20 mL去离子水,记为溶液B。混合溶液A与B并持续搅拌1 h后,将搅拌好的溶液转移至水热釜中在180 °C下加热12 h。将形成的固液混合物经抽滤分离,用去离子水洗涤固体直至为中性后,放入烘箱100 °C恒温12 h进行干燥。将干燥好的样品放入马弗炉以2 °C/min 升至500 °C 并保持3 h,形成CeθGa1-θOx载体。
CeθZr1-θOx载体的制备:称取2.7643 g NaOH 加入10 mL 去离子水中,记为溶液A。分别称取相应质量(n(Zr):n(Ce)=1:1)的硝酸氧锆水合物和六水合硝酸铈,并加入20 mL去离子水,记为溶液B。混合溶液A与B并持续搅拌1 h,其余步骤同上述载体的制备。
CeθFe1-θOx载体的制备:称取1.4374 g NaOH 加入3 mL去离子水中,记为溶液A。分别称取相应质量(n(Fe):n(Ce)=0.3:0.7)的九水合硝酸铁和六水合硝酸铈,并加入20 mL去离子水,记为溶液B。混合溶液A与B并持续搅拌1 h,其余步骤同上述载体的制备。
CeO2-Octa载体制备:称取2.7643 g NaOH与3.0000 g六水合硝酸铈分别加入10.09 mL和17.28 mL去离子水中,混合两种溶液并持续搅拌1 h。将搅拌好的溶液转移至水热釜中,之后放入烘箱180 °C 下保持12 h 进行水热处理。将形成的固液混合物进行抽滤,用去离子水洗涤固体直至pH为中性,之后放入烘箱100 °C 下保持12 h 进行干燥。将干燥好的样品放入马弗炉以2 °C/min升至500 °C并保持3 h,形成CeO2-Octa载体。
1.2.2 催化剂制备
催化剂采用浸渍法制备:称量0.1304 g 六水合硝酸镍溶于5 mL去离子水中,并称量0.5000 g制备好的载体(CeO2-Octa,CeθM1-θOx)加入上述镍盐溶液中,在60 °C搅拌直至搅干,然后将所得样品放置烘箱内80 °C干燥12 h。将干燥好的样品放入马弗炉400 °C 焙烧1.5 h,得到催化剂前驱体。对催化剂前驱体进行造粒(粒径180~250 μm),然后放入管式炉内400 °C 还原3 h,得到的催化剂分别记为Ni/CeO2-Octa与Ni/CeθM1-θOx。
1.3 催化剂表征
采用透射电子显微镜(FEⅠ Tecnai F20)对催化剂的形貌、尺寸进行分析。测试前将催化剂超声后在铜网上进行制样。
采用X 射线衍射仪(Rigaku Ultima Ⅳ)对催化剂的晶相结构进行分析。辐射源为Cu 靶Kα射线(λ=0.154 nm)。测试条件为:扫描范围10°~90°,扫描速率4 (°)/min。
采用X射线光电子能谱仪(AXⅠS Supra)对催化剂的表面结构及电子状态进行分析。称取一定量的催化剂进行压片,放置在400 °C 管式炉中还原3 h,然后在手套箱内进行样品的装填。
采用可见拉曼光谱仪(Renishaw inVia)对催化剂的表面氧空位相对含量进行测定。测试条件为:激发波长514 nm,波数范围200~1000 cm-1,功率10 MW。
采用化学吸附仪(Autochem ⅠⅠ-2920)进行H2程序升温还原(H2-TPR)与H2程序升温脱附(H2-TPD)分析。催化剂用量为50 mg。H2-TPR表征测试步骤为:400 °C 下在流动Ar 气氛中预处理60 min,然后降温至50 °C 切换气体为10% H2/Ar,以10 °C/min的升温速率升至1000 °C进行脱附。H2-TPD表征测试步骤为:400 °C下在H2中还原180 min,然后载气吹扫降温至50 °C。切换气体为10%H2/Ar 吸附120 min,然后He 气吹扫60 min,最后以10 °C/min的升温速率升至800 °C脱附。
1.4 催化剂催化性能评价
反应在200 mL的高压反应釜(安徽科幂仪器有限公司)中进行,反应条件为280 °C、6 MPa。称取一定质量的原料(w(正十二烷)=1%、w(DBF)=3%、w(正癸烷)=96%)加入反应釜内衬中,超声0.5 h使其充分混合,混合均匀后取0.5 mL样品作为反应的“00”点。加入60 mg筛分好的催化剂(粒径180~250 μm),进行装釜。对反应釜装置进行检漏步骤为:充入6 MPa 的氮气30 min 后观察压力表示数有无变化,若无变化则气密性良好,接着对其进行3次排空。最后,设置反应升温程序,在0.1 MPa氮气、700 r/min的条件下60 min 升温至反应温度(280 °C),取样记为“0”点,之后向反应釜内充入氢气反应180 min,在反应过程中每隔10 min进行取样并标记。
采用气质联用仪(Agilent-7890B/5977A)对反应过程中收集的样品进行定性及定量分析。DBF转化率(X,%)、产物选择性(S,%)及收率(Y,%)的计算公式分别见式(1)~式(3)。
式中,n0为反应初期DBF的物质的量,mol;n为反应完成时DBF 物质的量,mol;n产物为反应过程中产物的物质的量,mol。
2 结果与讨论
2.1 催化剂物相组成与形貌分析
不同载体的X 射线衍射(XRD)谱图如图1(a)所示。
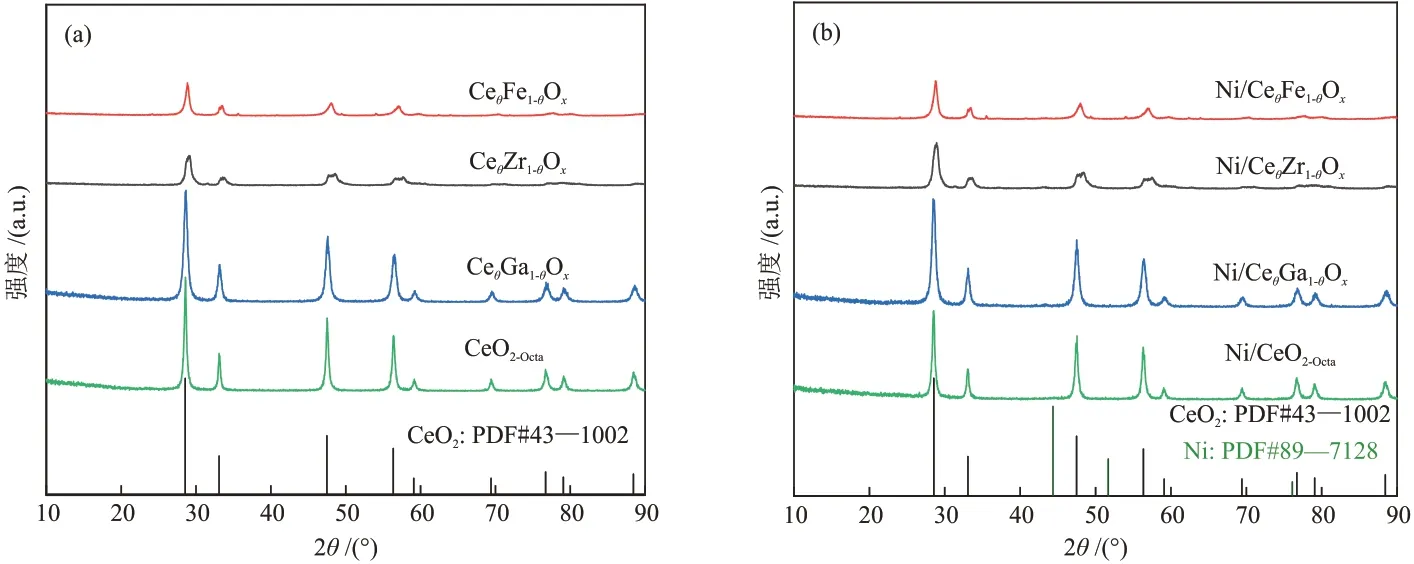
图1 不同载体(a)及催化剂(b)的XRD谱图Fig.1 XRD patterns of different supports (a) and catalysts (b)
由图1(a)可知,所有载体均为立方萤石结构[12],在2θ为28.5°、33.1°、47.4°和56.3°处存在衍射峰,分别对应CeO2的(111)、(100)、(110)和(200)晶面。CeO2掺杂Ga、Zr、Fe后未发现与掺杂相物种相关的衍射峰,表明金属阳离子进入CeO2晶格形成了固溶体结构,或其相关物种高度分散在CeO2表面[13]。不同催化剂的XRD 谱图见图1(b)。负载Ni后的催化剂显示出与载体相似的衍射峰,且没有关于NiO(111)与Ni(111)的新衍射峰出现,这归因于Ni物种在载体表面具有较高的分散度。此外,通过计算CeθM1-θOx载体的晶格参数,发 现CeθM1-θOx的载体晶格参数均小于CeO2的晶格参数(表1),证明离子半径大的Ce4+(r=0.092 nm)可以被离子半径小的金属离子取代,且不会引起晶体结构发生变化[14-15]。与未掺杂的CeO2相比,Ni掺杂后的催化剂的CeO2衍射峰变宽,表明其结晶度下降。由于低价态Mn+的掺杂置换了Ce4+,为了保持载体电中性,载体表面产生了氧空位。
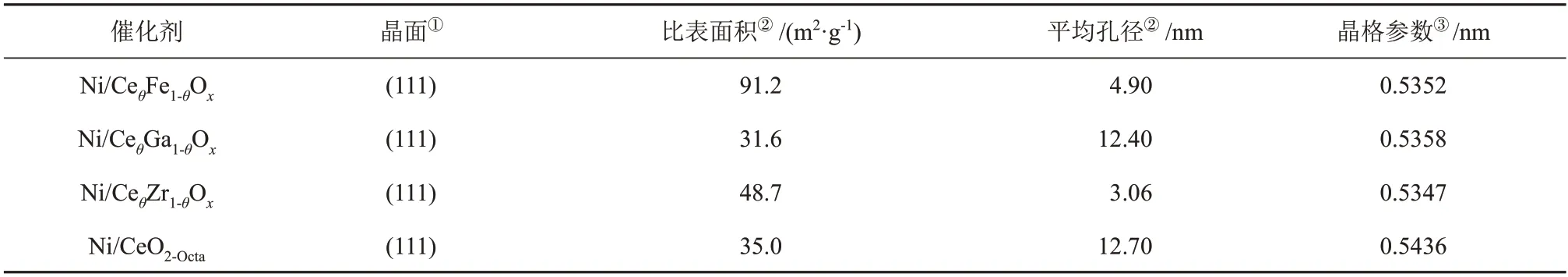
表1 不同催化剂的织构性质Table 1 Texture properties of different catalysts
Ni/CeO2-Octa和Ni/CeθM1-θOx(M=Ga,Zr,Fe)催化剂的形貌见图2。Ni/CeO2-Octa催化剂呈现纳米八面体的形貌,存在0.31 nm和的0.20 nm的晶格条纹,分别对应CeO2(111)和Ni(111)面。CeO2掺杂Ga、Zr后,其纳米八面体的形貌得以保留,但相比于未掺杂的Ni/CeO2-Octa催化剂,其形貌变得粗糙,CeO2掺杂Fe后呈现纳米多聚体的形貌。Ni/CeθM1-θOx催化剂均可得到属于Ni(111)和CeO2(111)两种晶格条纹,说明催化剂上暴露晶面为(111)。由表1 可知,Ni/CeθFe1-θOx催化剂的比表面积最大(91.2 m2/g),其次为Ni/CeθZr1-θOx催化剂的比表面积(48.7 m2/g),Ni/CeO2-Octa和Ni/CeθGa1-θOx催化剂的比表面积接近。标定催化剂选区电子衍射(SAED)照片中的电子衍射斑点,可以观察到Ni/CeO2-Octa和Ni/CeθM1-θOx催化剂中存在属于立方萤石结构的CeO2(111)、(100)、(110)、(311)和(222)面,与XRD结果一致。

图2 不同催化剂的TEM((a)~(d))、HRTEM((e)~(h))和SAED((i)~(l))照片Fig.2 TEM ((a)~(d)),HRTEM ((e)~(h)) and SAED ((i)~(l)) images of different catalysts
2.2 催化剂表面氧空位分析
2.2.1 X射线光电子能谱分析
利用XPS 表征定量分析催化剂表面氧空位相对含量,结果见图3。由图3(a)可知,在884.1 eV、897.9 eV 和902.7 eV 处的峰属于Ce3+的特征峰,其余5 种特征峰归属于Ce4+[16-17]。与Ni/CeO2-Octa相比,Ni/CeθM1-θOx催化剂中Ce的特征峰向高结合能移动,这是由于掺杂后金属离子之间置换引入了更多的氧空位。由于Ce4++O2-→Ce3++OV,因此Ce3+的原子比例(Ce3+/(Ce3++Ce4+))大代表催化剂表面形成了更多的氧空位。通过对比拟合出的Ce3+峰面积占Ce3++Ce4+峰面积的比例,可知Ce3+的原子比例的顺序由大到小为Ni/CeθFe1-θOx、Ni/CeθGa1-θOx、Ni/CeθZr1-θOx、Ni/CeO2-Octa。对O 1s 进行荷电校正分峰拟合,得到关于CeO2晶格氧(OL)与氧空位吸附氧物种(OV)的衍射峰[18]。Ni/CeθFe1-θOx催化剂在结合能为534.1 eV 处出现新的拟合峰,该峰归属于物理吸附氧物种(水和羟基等)[19]。催化剂表面氧空位相对含量可通过OV在O 中的占比(OV/(OL+OV))确定,由表2可知其大小顺序与Ce3+的原子比例一致,表明CeO2掺杂金属离子后会使晶体发生晶格畸变,导致催化剂表面产生更多的氧空位[20-21]。
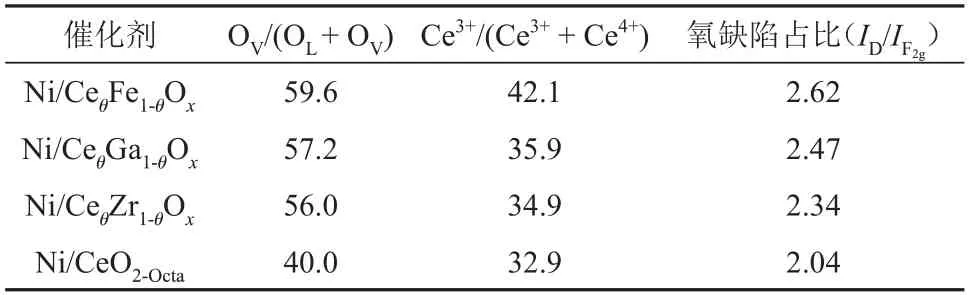
表2 不同催化剂的表面氧空位信息Table 2 Surface oxygen vacancy information of different catalysts
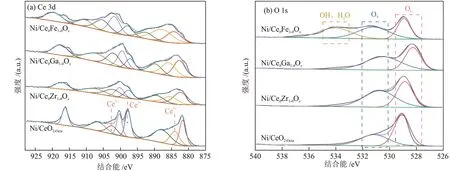
图3 不同催化剂的XPS谱图Fig.3 XPS patterns of different catalysts
2.2.2 拉曼光谱分析
利用可见拉曼光谱对比了Ni/CeO2-Octa和Ni/CeθM1-θOx催化剂表面氧空位的相对含量,结果见图4。
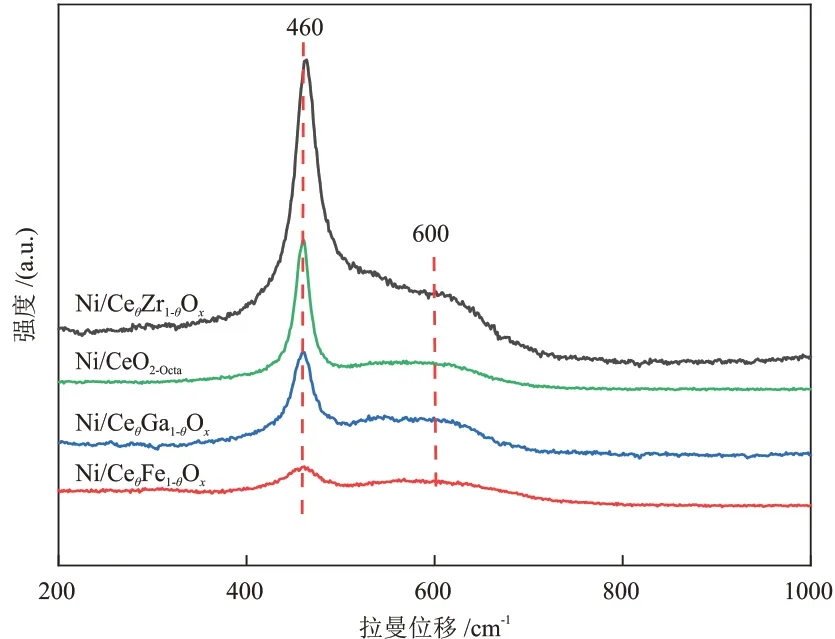
图4 不同催化剂的拉曼谱图Fig.4 Raman spectra of different catalysts
由图4 可知,Ni/CeO2-Octa和Ni/CeθM1-θOx催化剂存在两种特征峰,分别位于460 cm-1处(属于CeO2中Ce—O 伸缩振动的F2g峰)和600 cm-1处(属于CeO2中氧缺陷的D 峰)[22-23]。通过氧缺陷占比比较了催化剂表面氧空位的相对含量,由大到小依次为Ni/CeθFe1-θOx、Ni/CeθGa1-θOx、Ni/CeθZr1-θOx、Ni/CeO2-Octa,与XPS表征结果一致。
2.3 H2-TPR表征分析
CeθM1-θOx(M=Ga,Zr,Fe)和CeO2-Octa载体 的H2-TPR 表征结果见图5(a)。CeO2-Octa载体存在两个还原峰,分别属于表面氧的还原和晶格氧的还原,其中α峰为载体晶格氧的还原。由于金属离子掺杂后,载体上同时存在CeO2和掺杂金属氧化物[24]。CeθFe1-θOx载体在395 °C 和709 °C 处的还原峰归属于Fe氧化物的还原,509 °C处的还原峰为载体表面氧的还原[25],CeθZr1-θOx和CeθGa1-θOx载体的表面氧还原峰分别位于549 °C 和435 °C。CeθFe1-θOx、CeθGa1-θOx、CeθZr1-θOx和CeO2-Octa载体的耗氢量分别为0.104 mmol、0.044 mmol、0.053 mmol和0.035 mmol(表3)。
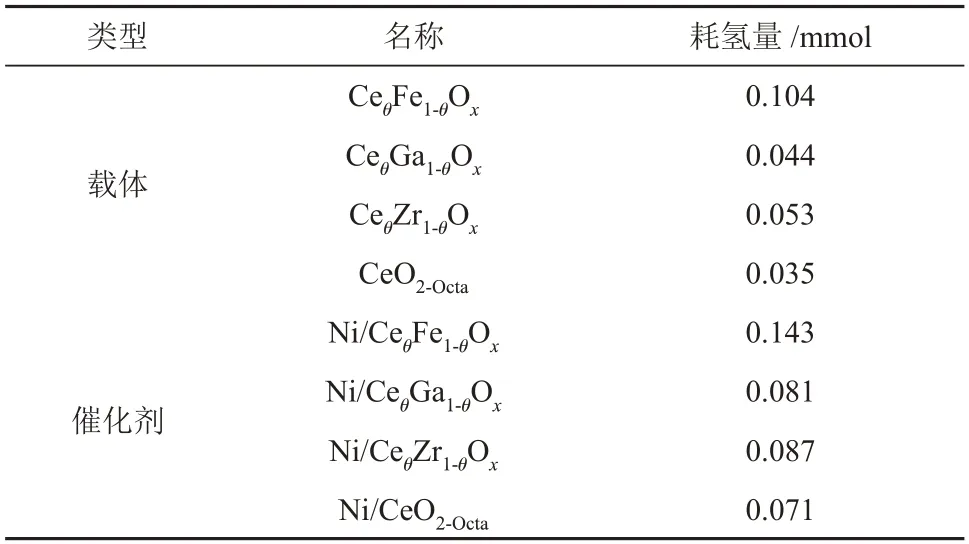
表3 不同载体和催化剂的耗氢量Table 3 Hydrogen consumption of different supports and catalysts
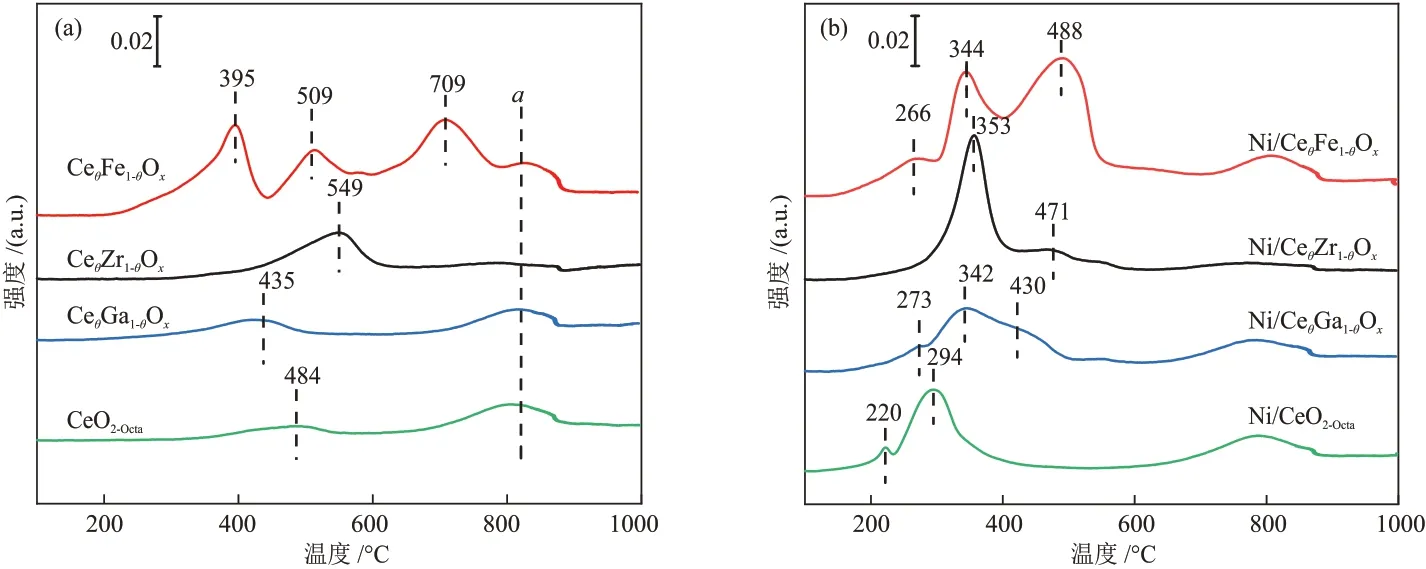
图5 不同载体(a)和催化剂(b)的H2-TPR曲线Fig.5 H2-TPR curves of different supports (a) and catalysts (b)
Ni/CeθM1-θOx和Ni/CeO2-Octa催化剂的H2-TPR 表征结果见图5(b)。理论上活性金属Ni 的耗氢量为0.042 mmol,Ni/CeθFe1-θOx、Ni/CeθGa1-θOx、Ni/CeθZr1-θOx和Ni/CeO2-Octa催化剂的耗氢量分别为0.143 mmol、0.081 mmol、0.087 mmol 和0.071 mmol,均大于理论上活性金属Ni 的耗氢量,说明Ni/CeθM1-θOx和Ni/CeO2-Octa催化剂上的还原峰除NiO的还原外还存在其他物质的还原。结合载体的还原峰对催化剂上的还原峰进行分析,Ni/CeO2-Octa催化剂在220 °C处的还原峰归属于Ni-CeO2-x固溶体产生氧空位处吸附氧的还原[22],在294 °C 处的还原峰归属于NiO 的还原和载体表面氧的还原,Ni/CeθGa1-θOx、Ni/CeθZr1-θOx催化剂在300~600 °C 处的还原峰归属于NiO的还原和载体表面氧的还原,Ni/CeθFe1-θOx催化剂在344 °C和488 °C 处的还原峰归属于NiO、载体表面氧和Fe3+的还原。金属Ni 的负载促进了载体的还原,使载体表面氧的还原温度降低。相比于Ni/CeO2-Octa催化剂,Ni/CeθM1-θOx催化剂还原峰位置向高温偏移,Ni/CeθM1-θOx催化剂中金属载体相互作用更强。
2.4 H2-TPD表征分析
不同催化剂的H2-TPD 曲线见图6。由图6 可知,在50~250 °C和250~400 °C处有3个H2脱附峰。α和β峰归属于H2在Ni颗粒上的吸附,γ峰归属于金属载体界面上H2的吸附[23]。对α和β峰面积进行积分,计算得到Ni/CeθFe1-θOx、Ni/CeθGa1-θOx、Ni/CeθZr1-θOx和Ni/CeO2-Octa催化剂的Ni 颗粒上H2的脱附量,分别为36.1 µmol/g、51.8 µmol/g、39.7 µmol/g和88.1 µmol/g。Ni/CeO2-Octa催化剂上H2脱附量最高,说明Ni/CeO2-Octa具有较小的Ni颗粒尺寸。高温下部分载体会与金属发生氢溢流,Ni/CeθFe1-θOx催化剂在446 °C处的脱附峰可归因于溢流氢物种的脱附[26]。
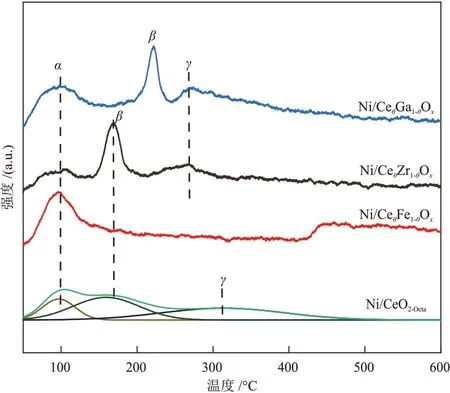
图6 不同催化剂的H2-TPD曲线Fig.6 H2-TPD curves of different catalysts
2.5 催化剂催化性能评价分析
Ni/CeO2-Octa催化剂催化DBF 的HDO 反应产物的产率变化见图7。反应过程中存在联环己烷(BCHs)、环己基环己烯(CHCHE)、环己基苯(CHB)和苯基环己烯(CHEB)4种脱氧产物,四氢二苯并呋喃(THDBF)、六氢二苯并呋喃(HHDBF)、十二氢二苯二呋喃(DHDBF)、环己基苯酚(CHPOH)、环己基环己醇(CHCHOH)和环己基环己酮(CHCHO)6 种含氧中间产物,以及裂解产物(CH)。由图7 可知,随着反应的进行,含氧中间产物产率与脱氧中间产物(CHCHE、CHB和CHEB)产率均呈现先增加后减小的趋势。CHCHOH 为反应过程中生成的主要含氧中间产物,BCHs为最终脱氧产物。
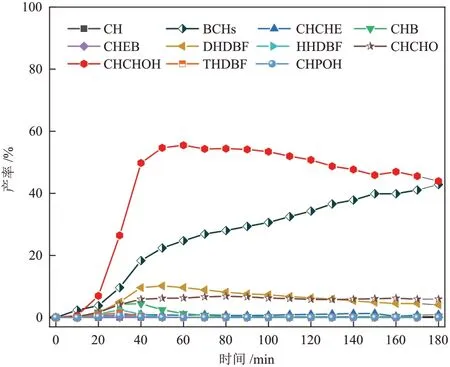
图7 Ni/CeO2-Octa 催化剂催化DBF 的HDO 反应产物的产率变化Fig.7 Changes in yields of products from HDO of DBF catalyzed by Ni/CeO2-Octa catalysts
以上分析可推断出DBF 的HDO 反应的路径[27-28],见图8。首先DBF 部分或全部加氢产生THDBF、HHDBF和DHDBF,然后HHDBF分别断裂sp3和sp2C—O 键,形 成CHPOH 和苯基环己醇(PCHOH),反应过程中PCHOH 未检出且CHEB 收率小于0.05%,表明sp2C—O 键断裂较难,不宜进行。生成的CHPOH 一部分氢化形成CHCHO,CHCHO生成CHCHOH;一部分断裂C—OH键形成CHB。完全加氢产物DHDBF 则通过断裂C—O 键生成CHCHOH,CHCHOH 发生脱水反应,形成CHCHE,CHCHE加氢产生完全加氢脱氧产物BCHs。
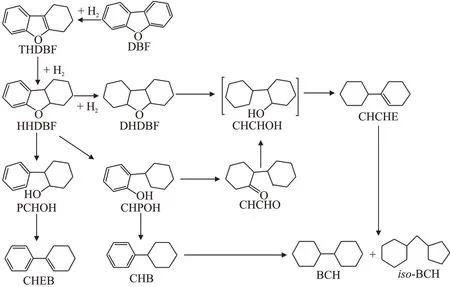
图8 DBF的HDO反应路径Fig.8 Reaction pathways of HDO of DBF
催化剂的XPS和拉曼表征结果表明催化剂表面氧空位相对含量由大到小的顺序为:Ni/CeθFe1-θOx、Ni/CeθGa1-θOx、Ni/CeθZr1-θOx、Ni/CeO2-Octa,为了明确表面氧空位相对含量对DBF 的HDO 反应的影响,对比了不同催化剂上产物的选择性,结果见图9。
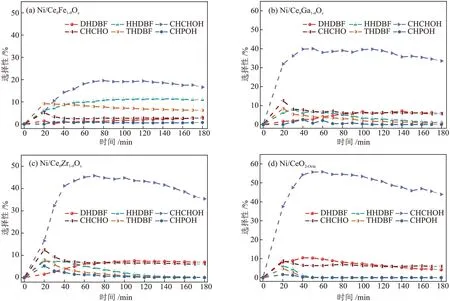
图9 反应过程中催化剂的不同含氧中间产物的选择性变化Fig.9 Changes of selectivity of different oxygen-containing intermediates of catalysts during reaction
由图9 可知,在4 种催化剂中均存在THDBF、HHDBF、DHDBF、CHPOH、CHCHOH 和CHCHO 6 种含氧中间产物,其中CHCHOH 的选择性最高。在反应60 min 后,CHCHOH 的选择性基本都达到最高,而后随着反应的进行,CHCHOH 选择性逐渐降 低。反 应180 min 时,Ni/CeO2-Octa、Ni/CeθFe1-θOx、Ni/CeθGa1-θOx和Ni/CeθZr1-θOx催化剂的CHCHOH 的选择性分别为42.93%、16.57%、33.55%和35.38%,Ni/CeθM1-θOx催化剂的CHCHOH 的选择性均低于Ni/CeO2-Octa催化剂的CHCHOH 的的选择性。该结果表明氧空位相对含量较少的Ni/CeO2-Octa催化剂的CHCHOH的转化相对缓慢。
反应结束后Ni/CeO2-Octa和Ni/CeθM1-θOx催 化剂的含氧中间产物的选择性见图10。
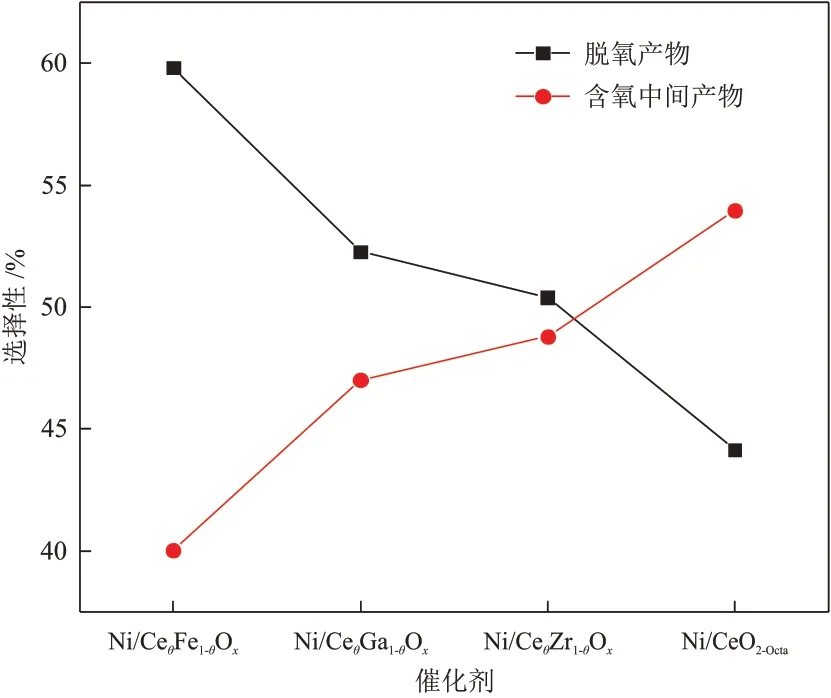
图10 不同催化剂的产物的选择性对比Fig.10 Comparison of product selectivity of different catalysts
由图10可知,Ni/CeO2-Octa、Ni/CeθFe1-θOx、Ni/CeθGa1-θOx和Ni/CeθZr1-θOx催化剂的含氧中间产物的选择性分别为52.96%、40.03%、47.01%和48.79%,不同催化剂的含氧中间产物选择性的顺序由小到大为Ni/CeθFe1-θOx、Ni/CeθGa1-θOx、Ni/CeθZr1-θOx、Ni/CeO2-Octa。由此可知,掺杂后的Ni/CeθM1-θOx催化剂的催化活性更高,有利于含氧产物的进一步转化。
Ni/CeO2-Octa和Ni/CeθM1-θOx催化剂的脱氧产物的选择性见图11,脱氧产物包括BCHs、CHCHE、CHB和CHEB。4 种催化剂的CHEB 的选择性均低于0.50%,在反应20 min 时,4 种催化剂的CHCHE 和CHB的选择性达到最高,之后逐渐下降。整个过程中主要的脱氧产物为BCHs,其选择性呈现出持续上升的趋势。反应180 min时,Ni/CeO2-Octa、Ni/CeθFe1-θOx、Ni/CeθGa1-θOx和Ni/CeθZr1-θOx催化剂的BCHs 选择性分别为42.83%、40.14%、39.65%和40.03%,4 种催化剂的BCHs 的选择性差值<5.00%。反应结束后,催化剂的脱氧产物的选择性的顺序由大到小为Ni/CeθFe1-θOx(59.81%)、Ni/CeθGa1-θOx(52.27%)、Ni/CeθZr1-θOx(50.41%)、Ni/CeO2-Octa(44.15%),说明DBF 在Ni/CeθFe1-θOx催化剂上具有更优越的脱氧性能,在Ni/CeO2-Octa催化剂上的脱氧能力较弱。由于金属掺杂Ni/CeO2后会发生金属离子的置换,导致晶格畸变,催化剂表面氧空位增加,结合不同催化剂的脱氧产物的选择性可知,催化剂表面氧空位的增加有利于含氧中间产物的吸附和活化,促进了反应中C—O键的断裂,提高了脱氧产物的选择性。
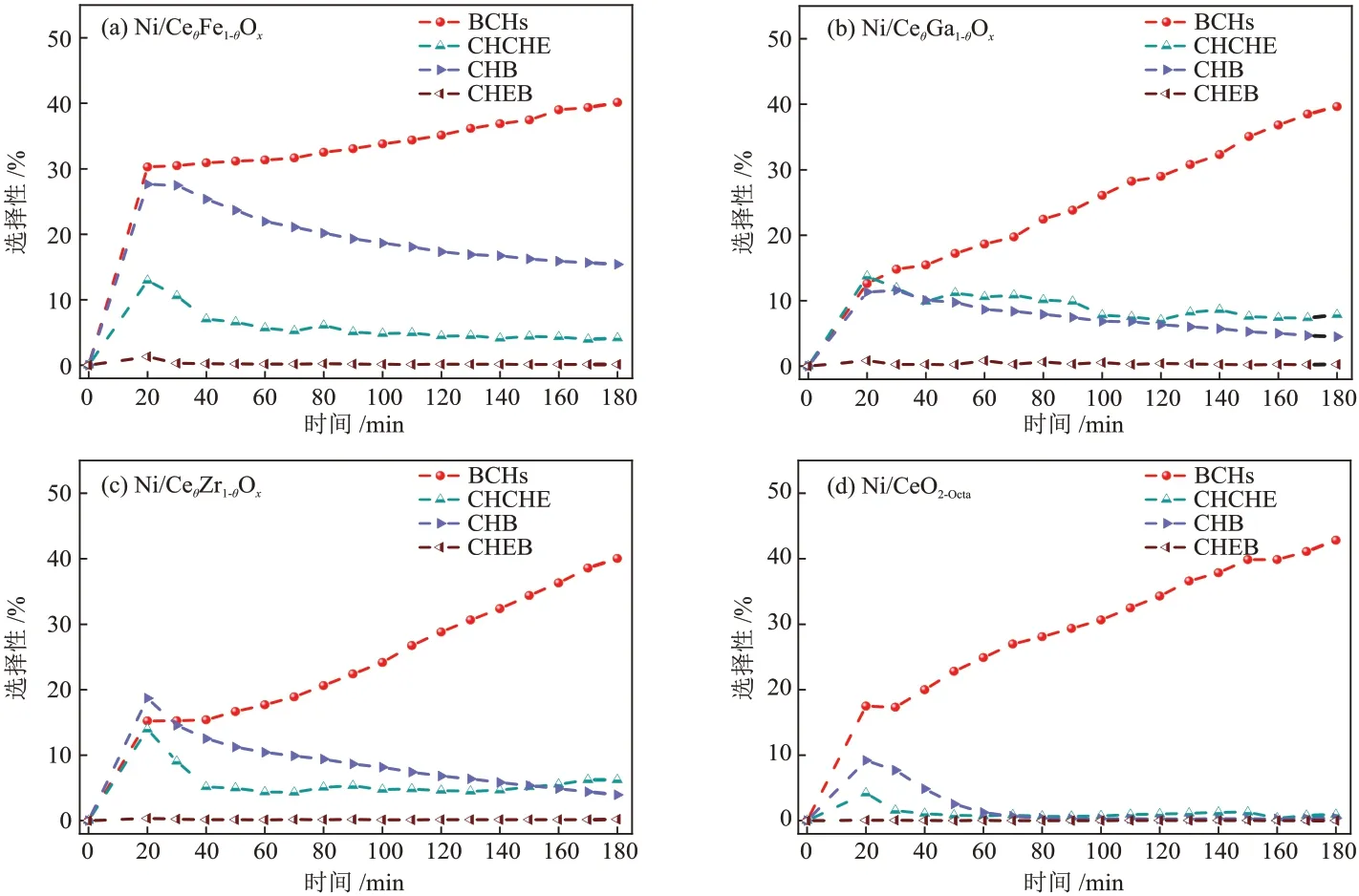
图11 反应过程中催化剂的脱氧产物的选择性变化Fig.11 Comparison of selectivity of deoxidation products of catalysts during reaction
3 结论
本文采用水热法制备了CeθM1-θOx和CeO2-Octa载体,采用浸渍法制备了Ni/CeθM1-θOx和Ni/CeO2-Octa催化剂,通过金属离子掺杂载体对催化剂表面氧空位进行了调控,研究了催化剂表面氧空位对DBF 的HDO反应的影响,得出如下结论。
(1)金属离子(Fe、Ga 和Zr)的掺杂有效地提高了Ni/CeθM1-θOx催化剂中表面氧空位的相对含量,氧空位的相对含量顺序由大到小为Ni/CeθFe1-θOx、Ni/CeθGa1-θOx、Ni/CeθZr1-θOx、Ni/CeO2-Octa。
(2)相比于Ni/CeO2-Octa催化剂,Ni/CeθM1-θOx催化剂表面氧空位相对含量高,金属载体相互作用增强。DBF的HDO反应180 min时,表面氧空位相对含量最高的Ni/CeθFe1-θOx催化剂上含氧中间产物的选择性最低,脱氧产物的选择性最高(59.81%)。催化剂表面氧空位相对含量增大,促进了反应物分子在催化剂上的吸附,有利于C—O 键的断裂生成脱氧产物,提高了催化剂的反应性能。
猜你喜欢
中学化学(2022年5期)2022-06-17
氯碱工业(2021年6期)2021-12-25
石油化工应用(2020年7期)2020-08-08
陶瓷学报(2019年5期)2019-01-12
中学生数理化·高二版(2016年3期)2016-12-26
中学生数理化·高二版(2016年3期)2016-12-26
玻璃纤维(2016年2期)2016-12-18
河南大学学报(医学版)(2015年1期)2015-03-17
读者欣赏(2014年6期)2014-07-03
语文知识(2014年2期)2014-02-28
