
棕榈酸调控circ_DENND4C 抑制非小细胞肺癌发生发展的作用机制研究
2023-12-12 02:53李佳维徐涛俞万钧吕佳佩
浙江医学 2023年22期
李佳维 徐涛 俞万钧 吕佳佩
肺癌是目前世界上发病率和死亡率最高的恶性肿瘤[1],其中非小细胞肺癌(non-small cell lung cancer,NSCLC)约占肺癌总数的80%[2-3]。环状RNA(circular RNAs,circRNAs)作为一种非编码RNA,能通过调节内源竞争RNA(competing endogenous RNA,ceRNA)或miRNA,调节基因转录,与其他蛋白质相互作用影响恶性肿瘤的发生、发展[4-5]。circRNA 的5'帽端和3'尾端形成特殊闭环结构对核酸外切酶具有内在的抵抗力[6],可能是更准确和有效的新型生物标志物。研究证实circRNAs 参与肺癌发生、发展的调控,例如has_circ_0000190 为多种促致癌信号通路的介质,可以靶向抑制肺癌进展[7];circ_NDUFB2 通过破坏IGF2BPs 和激活抗肿瘤免疫来抑制NSCLC 的进展[8]。
脂质代谢重编程会影响细胞功能,从而导致癌变[9]。棕榈酸(C16:0,即含16 个碳原子的饱和脂肪酸)是动物中最丰富的饱和脂肪酸,可以作为其他脂质合成和β 氧化反应的底物。研究发现C16:0 可能参与黑色素瘤细胞转移[10],且与肝癌细胞转移能力成正相关[11]。研究表明circRNAs 在脂质代谢中起重要作用[12],但脂质如何影响circRNAs 的改变仍鲜有报道。
本团队前期研究发现NSCLC 患者血清中C16:0 含量显著降低,通过体内及体外实验证实C16:0 能显著抑制NSCLC 细胞增殖、迁移及侵袭,诱导细胞凋亡[13-14]。为进一步探究C16:0 抑癌机制,本研究构建C16:0 刺激细胞模型,通过circRNAs 高通量测序,筛选出肿瘤相关因子环状含有DENN 结构域蛋白4C(circ_DENN domain-containing protein 4C,circ_DENND4C)。本研究结合脂质及circRNAs 组学,分析C16:0 抑制A549 细胞增殖、迁移和侵袭能力的作用及机制,为NSCLC 的治疗提供了新靶点与思路。
1 材料和方法
1.1 细胞、主要试剂和仪器 人NSCLC 细胞系A549(美国模式培养物集存库);DMEM 培养基(美国Gibco公司,规格:500 mL,批号:C11995500BT);10%FBS(以色列BI 公司,批号:04001-1A);100 U/mL 青霉素和100 μg/mL 链霉素(美国Gibco 公司,100X,批号:15070063);0.25%胰蛋白酶(美国Gibco 公司,批号:25200114);Opti-MEM 培养基(美国Gibco 公司,批号:10370070);C16:0(德国Sigma 公司,批号:SLCC6727);circRNAs 测序(中国上海美吉生物公司医药科技有限公司);Trizol 试剂(美国SIGMA 公司,批号:T9424);TransScript First strand cDNA Synthesis SuperMix for PCR(北京全式金生物技术股份有限公司,批号:AU34-02);7500 实时荧光定量PCR 仪(美国Applied Biosystems 公司);PerfectStart Green qPCR SuperMix(北京全式金生物技术股份有限公司,批号:AQ602-21);circ_DENND4C 过表达载体(上海GenePharma 公司);结晶紫(国药集团化学试剂有限公司,批号C805211);Lipofectamine 2000(美国Thermo Fisher Scientific 公司,批号:11668030);TransDetect@Cell 计数套件(北京Trans 公司);微板读取器(美国cMax plus 公司);Image J 软件(美国国立卫生研究院);Matrigel 基质胶(北京索莱宝科技有限公司,批号:356234);光学显微镜(日本尼康公司)。
1.2 细胞培养 A549细胞接种于含10%FBS、100 U/mL青霉素和100 μg/mL 链霉素的DMEM 培养基中培养,置于37 ℃、5%CO2培养箱中培养。培养基每3 d更换1次。使用0.25%胰蛋白酶对细胞进行消化以传代培养。
1.3 细胞测序分组和造模 将A549 细胞分为对照组和C16:0 组。C16:0 组为circRNAs 测序实验组,采用C16:0 200 μmol/L 刺激A549 细胞;未做处理的A549 细胞作为对照组。每组3 个样本,48 h 后提取总RNA,样本RNA 送检用于高通量circRNAs 转录组测序。
1.4 circRNAs 表达水平检测 采用qRT-PCR 法。使用Trizol 试剂裂解细胞提取总RNA,提取的总RNA 保存在4 ℃冰箱以备后续的反转录及实时定量PCR 使用。使用TransScript First strand cDNA Synthesis Super-Mix for PCR 将1 μg 总RNA 反转录成cDNA,反应条件如下:37 ℃15 min,85 ℃5 s,4 ℃保存。用Perfect-Start Green qPCR SuperMix 在实时PCR 系统上进行qRT-PCR,反应条件如下:94 ℃30 s,94 ℃5 s,60 ℃15 s,72 ℃10 s,共40 个循环,所有实验重复3 次。以β-actin 为内参,采用2-ΔΔCt法计算circRNAs 相对表达水平。RT-PCR 引物序列见表1。

表1 qRT-PCR 引物序列
1.5 细胞转染及细胞功能实验分组 将A549 细胞接种至6 孔板中,每孔5×105个细胞,培养24 h,细胞生长至90%融合后,采用Lipofectamine 2000 进行细胞转染。分别将Lipofectamine 2000 和核苷酸加入到250 μL Opti-MEM 培养基中稀释,将稀释液混合,轻轻混匀,室温静置24 h 后,收集细胞,通过qRT-PCR 法检测细胞转染效率。将细胞分为空白对照组(NC 组)、C16:0组、circ_DENND4C 质粒过表达组(pex-0086466 组)、circ_DENND4C 质粒过表达联合C16:0 组(pex-0086466+C16:0 组)进行细胞功能实验。
1.6 细胞增殖能力检测 采用细胞计数试剂盒(cell counting kit-8,CCK-8)法。将转染的A549 细胞转移到96 孔板,每孔2×103个细胞,培养24 h,待细胞贴壁后更换为DMEM 培养基,饥饿培养24 h 后定义为第0 天,此时更换为含有10% FBS、100 U/mL青霉素和100 μg/mL链霉素的DMEM 培养基。分别于第0、1、2 天在C16:0组及pex-0086466+C16:0 组中加入C16:0(4 μL/孔,浓度10 mmol/L),继续培养。第3 天,向每个孔中加入10 μL CCK-8 试剂+90 μL DMEM 培养基。使用酶标仪检测450 nm 波长处吸光度(optical density,OD)值。细胞增殖率计算公式:细胞增殖率(%)=(OD实验组-OD空白组)/(OD对照组-OD空白组)×100%。
1.7 细胞菌落形成能力检测 采用细胞克隆形成试验。将转染的A549 细胞转移到6 孔板,每孔500 个细胞,在C16:0 组及pex-0086466+C16:0 组中加入C16:0(40 μL/孔,浓度10 mmol/L),培养2 周,每3~4 d 更换1 次培养基。然后弃掉孔中培养基,用甲醛固定菌落20 min,用0.5%结晶紫溶液染色20 min。PBS 冲洗3 次后,用肉眼计数染色的菌落。菌落形成率=菌落数/1 000×100%。
1.8 细胞迁移能力检测 采用细胞划痕试验。将转染的A549 细胞接种至6 孔板中,每孔5×105个细胞,恒温培养箱培育24 h。用200 μL 无菌移液管尖端(记录0 h)创建清晰的无细胞区,用PBS 清洗细胞3 次,去除划下的细胞,使用无血清培养基培养;在C16:0 组及pex-0086466+C16:0 组中加入C16:0(40 μL/孔,浓度10 mmol/L),放入37 ℃、5% CO2培养箱培养48 h,分别在0、24、48 h 使用光学显微镜拍摄细胞迁移距离。迁移细胞数和初始接种细胞数的比值为相对迁移细胞数(%)。
1.9 细胞侵袭能力检测 采用Transwell 侵袭实验。使用Matrigel 涂覆Transwell 小室底部膜的上室,至37 ℃30 min 使Matrigel 聚合成凝胶。将转染的细胞用无血清培养基重新悬浮,转移到Transwell 小室上室中,每孔5×104个细胞,在下室中加入500 μL 含有10%FBS的培养基。细胞培养48 h,取出Transwell小室,弃掉孔中培养基,用甲醇固定穿过膜的细胞20 min后风干,PBS 清洗3 遍,随后用0.1%结晶紫溶液染色15 min,染色后用PBS 反复清洗残余的结晶紫。最后在光学显微镜(×200)下随机选取10 个视野/孔计数细胞。侵袭细胞数和初始接种细胞数的比值作为相对侵袭细胞数(%)。
1.10 统计学处理 采用SPSS 17.0 统计软件、Graphpad Prism 9.0 统计软件。计量资料以表示,两组间比较采用两独立样本t检验;多组间比较采用单因素方差分析,两两比较采用LSD-t检验。P<0.05 为差异有统计学意义。
2 结果
2.1 C16:0 对NSCLC 细胞中circRNAs 组学的影响C16:0刺激A549细胞48 h后高通量测序检测到2 260个circRNAs(图1A,见插页)。发现C16:0 可以引起A549细胞内circRNAs 表达改变,其中有意义的上调36 个,下调227 个(P<0.05)(图1B,见插页)。因此设想C16:0可能调控circRNAs 影响NSCLC 发生、发展。从中挑选出上调及下调变化前10的circRNAs(图1C,见插页),这些circRNAs表达改变均在对照组5倍以上(图2)。
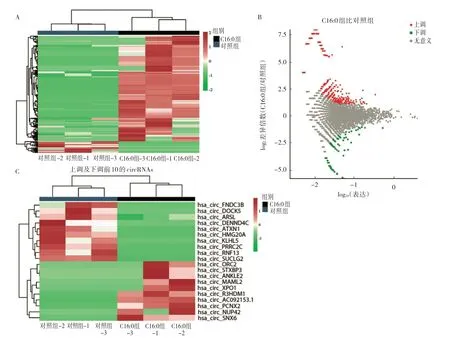
图1 C16:0 处理后的A549 细胞测序结果分析(A:circRNAs 表达变化的层次聚类分析图,红色和绿色分别表示高表达和低表达;B:circRNAs 表达变化的火山图,红点和绿点分别代表上调及下调>2 倍的circRNAs;C:表达上调及下调前10 的circRNAs 层次聚类分析图)
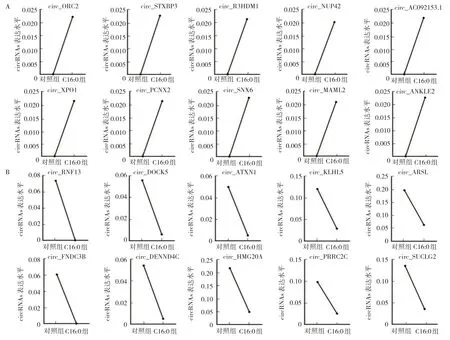
图2 C16:0 处理后的A549 细胞测序结果分析(A:表达上调前10 的circRNAs 测序结果;B:表达下调前10 的circRNAs 测序结果)
2.2 验证C16:0 相关的circRNAs 表达 结合上述测序结果中上调及下调变化前10 的circRNAs 及circinteractome 数据库(https://circinteractome.nia.nih.gov/index.html)、人类circRNAs 数据库(http://www.circbase.org/)中肿瘤相关circRNAs 的结果,取交集得到14 个(7 个表达上调,7 个表达下调)可能与C16:0 影响NSCLC 生长有关的circRNAs。通过qRT-PCR 验证,结果发现上调因子中circ_PCNX2、circ_ORC2、circ_NUP42、circ_XPO1表达变化与测序结果一致,但差异均无统计学意义(均P>0.05)(图3A);下调因子中circ_DENND4C、circ_ARSL、circ_DOCK5 表达变化与测序结果一致,其中circ_DENND4C 加药后表达下调近5 倍,差异有统计学意义(P<0.05)(图3B)。因此认为C16:0 可能通过抑制circ_DENND4C表达从而达到抑制NSCLC的生长。
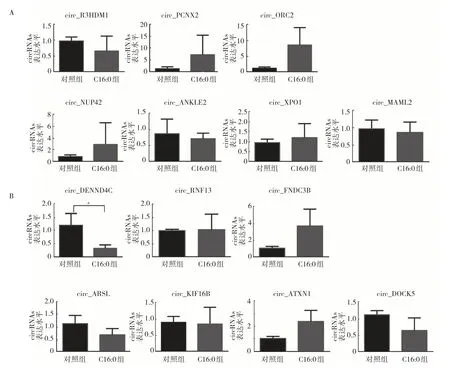
图3 C16:0 调控肿瘤相关circRNAs 表达(A:qRT-PCR 验证在C16:0 加药后测序结果显示表达上调前10 且与肿瘤相关的7 个circRNAs;B:qRT-PCR 验证在C16:0 加药后测序结果显示表达下调前10 且与肿瘤相关的7 个circRNAs,与对照组比较,aP<0.05)
2.3 C16:0 通过调控circ_DENND4C 对NSCLC 细胞增殖、菌落形成、侵袭和迁移的影响 为了验证circ_DENND4C 在NSCLC 中的生物学功能及其在C16:0 抑制NSCLC 发生、发展中的作用,在A549 细胞中通过质粒介导circ_DENND4C 过表达,然后利用C16:0 刺激A549 细胞,采用qRT-PCR 验证circ_DENND4C 的mRNA 表达情况,C16:0 组中circ_DENND4C 表达下调,pex_0086466 组中circ_DENND4C 表达上调,而NC 组与pex-0086466+C16:0 组中circ_DENND4C 表达量比较差异无统计学意义(P>0.05)(图4A,见插页)。利用CCK-8与克隆试验发现,circ_DENND4C 过表达可以促进A549 细胞的增殖和克隆能力,C16:0 可以抑制A549细胞的增殖和克隆能力,而过表达circ_DENND4C 可以逆转C16:0 对A549 细胞的抑制作用(图4B-C,见插页)。划痕试验表明,过表达circ_DENND4C 可以使A549 细胞的迁移能力大幅上升,C16:0 可以抑制其迁移,而过表达circ_ DENND4C 可以逆转C16:0 对A549细胞迁移能力的抑制(图4D,见插页)。Transwell 侵袭实验表明,pex_0086466 组细胞侵袭率约为NC 组的2倍,C16:0 组细胞侵袭率仅有NC 组的30%,而过表达circ_DENND4C 后加入C16:0 细胞侵袭率与NC 组相似(图4E,见插页)。以上实验说明circ_DENND4C 可以促进NSCLC 增殖、侵袭和迁移,是C16:0 抑制NSCLC 的关键因子。
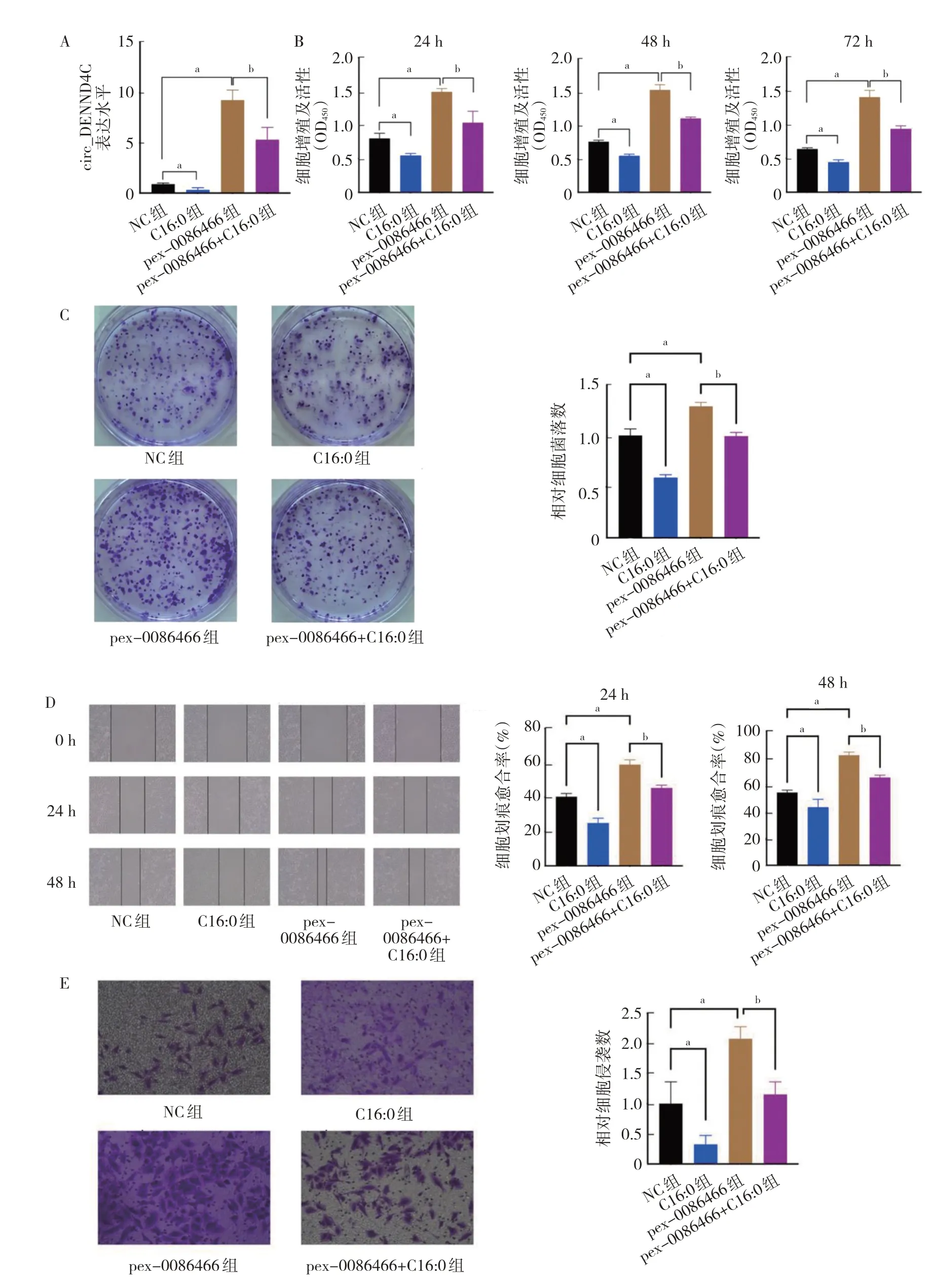
图4 C16:0 通过调控circ_DENND4C 对A549 细胞增殖、集落形成、侵袭和迁移的影响(A:4 组A549 细胞中circ_DENND4C 表达水平比较;B:4 组A549 细胞增殖能力比较;C:4 组A549 细胞的克隆数比较;D:4 组A549 细胞迁移能力比较;E:4 组A549 细胞侵袭能力比较,结晶紫染色,×200)
3 讨论
NSCLC 是呼吸系统常见的恶性肿瘤之一,与小细胞肺癌相比,NSCLC 癌细胞的生长和分裂更慢,传播和转移相对较晚,约占所有肺癌总数的80%~85%,大部分患者确诊时已经处于中晚期,5 年生存率低于54%[1]。因此,发现治疗NSCLC 的新型分子靶点,探索其中的调控通路是急需解决的问题。尽管人们长期以来一直认为脂质在癌症中起着关键作用,但特征变化的程度和复杂性及其在癌症和生理学中的作用直到现在才被曝光。近年来,基于液相色谱-质谱技术的高速发展,已经发现了许多关于癌症脂质代谢的变化[15]。本团队前期研究发现NSCLC 患者血浆中脂质谱与健康志愿者具有明显异质性,并筛选出一系列相关脂质组群,其中NSCLC 患者血浆中C16:0 浓度明显低于健康志愿者。并进一步通过细胞功能实验发现C16:0 可以抑制NSCLC 的发生、发展[14],但其具体机制仍是未知。
目前越来越多的研究表明,circRNAs 与各类癌症的发生、发展密切相关[16]。 circRNAs 较线性RNA 相比,其在组织中更稳定,寿命更长,对核糖核酸酶更具有抵抗力[17],是一种潜在的生物标志物[12]。已有大量研究证实circRNAs 的异常表达与NSCLC 的发生、发展有关,例如,has-circ-100395 通过调节miR-1228/TCF21 途径来调节NSCLC 的增殖、迁移和侵袭功能[18];circ-001278 在肺腺癌细胞中表达显著上调,其可以促进肺腺癌组织中程序性死亡配体1/程序性死亡受体1的表达[19];circ_PTPRM 通过调节miR-139-5p/SETD5来促进NSCLC 的进展[20]。部分circRNAs 可以调节糖酵解和β 氧化参与肿瘤脂质代谢过程,Li 等[21]在肿瘤异种移植模型中,沉默或强制表达circ_ACC1 分别导致肿瘤生长抑制和增强,并提出了circ_ACC1 诱导AMP 依赖的蛋白激酶激活致肿瘤细胞在代谢重编程的观点。因此肿瘤脂质代谢过程与circRNAs 有着不可忽视的关联。
本研究通过circRNAs 测序发现circ_DENND4C 也许是C16:0 抑制NSCLC 发生、发展的关键因子。circ_DENND4C(has-circ-0086466)来源于DENND4C 基因外显子,是一种在肿瘤增殖中具有调节功能的缺氧相关分子。有研究表明,circ_DENND4C 在乳腺癌组织中高表达,沉默缺氧诱导因子-1 后其表达下调,且抑制缺氧下乳腺癌细胞的增殖[22]。circ_DENND4C 在脑胶质瘤中高表达,敲低circ_DENND4C 可破坏血肿瘤屏障(blood-tumor barrier,BTB)的完整性且提高渗透率,从而促进抗肿瘤药物阿霉素穿过BTB 诱导胶质瘤细胞凋亡[23]。circ_DENND4C 在肝癌细胞中也过度表达。此外circ_DENND4C 可以增强转录因子4 的表达,通过隔离miR-195-5p 激活Wnt/β-catenin 信号传导通路在肝癌细胞中发挥促肿瘤功能[24]。但目前尚未有关于circ_DENND4C 和NSCLC 的研究报道。本研究发现C16:0 可以抑制A549 细胞的增殖生长,同时抑制circ_DENND4C 的表达。当circ_DENND4C 过表达时,C16:0 对A549 细胞的抑制作用消失,且circ_DENND4C本身过表达可以促进A549 细胞增殖、迁移和侵袭能力。因此,本研究认为C16:0 通过调控促癌因子circ_DENND4C 表达从而抑制NSCLC 的发生、发展。本研究首次整合了脂质质谱分析及circRNAs 测序双组学结果,从全新角度为C16:0 用于NSCLC 治疗奠定基础。
综上所述,circ_DENND4C 过表达可以促进NSCLC的进展,C16:0 可以通过下调circ_DENND4C 的表达抑制NSCLC 的发生、发展。这一发现为C16:0 治疗NSCLC 提供重要的实验依据。
猜你喜欢
航天电子对抗(2022年4期)2022-10-24
新民周刊(2022年27期)2022-08-01
传染病信息(2021年6期)2021-02-12
中国科技纵横(2018年2期)2018-11-29
中成药(2018年9期)2018-10-09
中成药(2018年7期)2018-08-04
中成药(2018年1期)2018-02-02
中成药(2017年4期)2017-05-17
生物医学工程学进展(2015年1期)2015-02-28
化学工业与工程(2015年1期)2015-02-10
