
6-氮杂脲嘧啶类化合物的合成与修饰研究进展
2023-11-22 02:28冯冉冉赵继全张宏宇
精细石油化工 2023年6期
冯冉冉,赵继全,张宏宇*
(1.河北工业大学化工学院,天津 300401;2.河北省绿色化工与高效节能重点实验室,天津 300401;3.天津市本质安全化工技术重点实验室,天津 300401)
6-氮杂脲嘧啶又名1,2,4-三嗪-3,5(2H, 4H)-二酮,由尿嘧啶6号位碳原子被氮原子取代而来,是尿嘧啶的一种重要的类似物。6-氮杂脲嘧啶骨架是多种生物活性分子的基本结构单元,基于6-氮杂脲嘧啶骨架修饰而得的氮杂脲嘧啶类化合物具有特殊的生理活性,表现出抗病毒、抗菌及抗肿瘤等活性[1-2]。相关药物包括用于治疗球虫病的抗球虫兽药地克珠利[3]、治疗精神分裂症的D-氨基酸氧化酶抑制剂[4]以及治疗癫痫的抗惊厥药[5]。特别是含6-氮杂脲嘧啶骨架的核苷类似物药物具有更好的抗病毒活性,如用于临床治疗致病性虫媒病毒的6-氮杂尿苷[6]、治疗艾滋病的强效双功能核苷探针[7],以及分枝杆菌胸苷酸合酶抑制剂[8](见图1)。因此,6-氮杂脲嘧啶类化合物受到人们的广泛关注,发现了多种合成与修饰6-氮杂脲嘧啶骨架以获得其衍生物的方法。笔者综述了6-氮杂脲嘧啶骨架合成与修饰的研究进展,为开发研究此类化合物提供借鉴。

图1 几种代表性6-氮杂脲嘧啶类药物分子
1 6-氮杂脲嘧啶骨架的合成方法
6-氮杂脲嘧啶类化合物具有特殊的生理和药理活性。6-氮杂脲嘧啶的经典合成是通过环化反应实现的。1947年,Walter[9]以氨基脲(1)与乙醛酸(2)为起始原料,首次实现6-氮杂脲嘧啶的人工合成(见图2)。该法虽然路线较短,原子利用率高,但是反应条件苛刻,副产物多,产率较低。
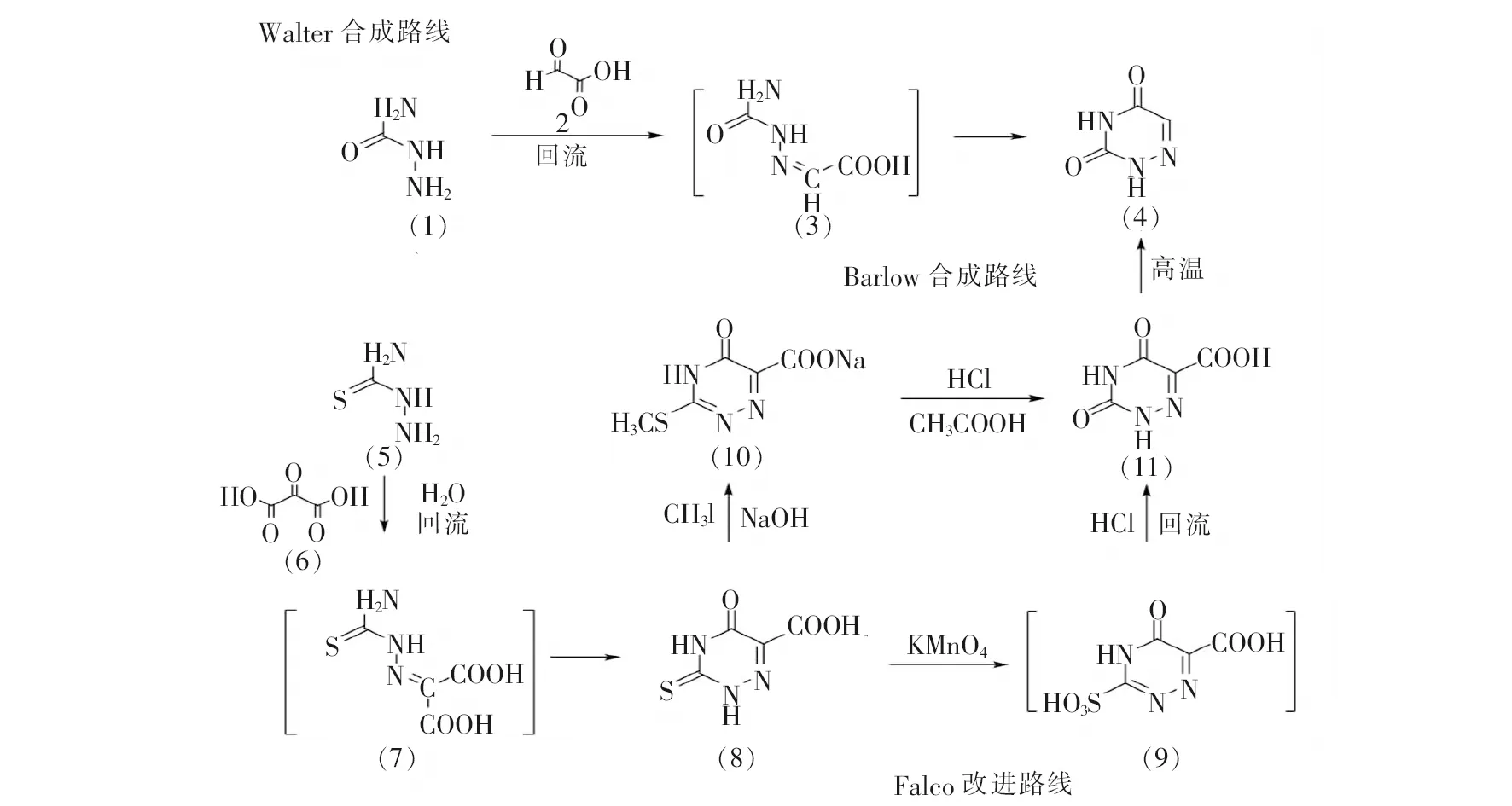
图2 6-氮杂脲嘧啶的早期合成方法
1955年,Barlow等[10]报道了一种新的合成路线。他们以氧代丙二酸(6)替代乙醛酸与氨基硫脲进行缩合反应,再经过环化得到5-氧代-3-硫代-1,2,4-三嗪-6-羧酸(7),而后经过甲基化及水解反应生成3,5-二氧代-1,2,4-三嗪-6-羧酸(11),最后在230 ℃的高温下发生脱羧反应并升华得到6-氮杂脲嘧啶(4)。该法虽然步骤较多,但除甲基化(77%)和甲基硫醚水解(55%)产率较低外,其他步反应产率均较高,总产率尚可。1956年,Falco等[11]对此合成路线进行了改进,避开甲基化和硫醚水解步骤,改用高锰酸钾将5-氧代-3-硫代-1,2,4-三嗪-6-羧酸氧化为5-氧代-3-磺基-1,2,4-三嗪-6-羧酸(9),再原位去磺化以较高的产率生成3,5-二氧代-1,2,4-三嗪-6-羧酸(11)(两步累积收率78%),使得总产率显著提高。
早期的合成路线分别使用乙醛酸与氨基脲,或氧代丙二酸与氨基硫脲作为起始原料,经过缩合反应、环化反应和脱羧反应等步骤得到6-氮杂脲嘧啶,存在过程繁琐和总体效率不高的缺点。因此,人们开发了更高效的合成方法。2016年,Bakavoli等[12]报道了一种以水合三氯乙醛(12)代替乙醛酸与氨基脲反应合成6-氮杂脲嘧啶的方法,见图3。其中,水合三氯乙醛与氨基脲经过缩合、水解等反应生成2-(2-氨基甲酰基肼基)乙酸中间体(3),该中间体再经分子内环化反应最终生成6-氮杂脲嘧啶,反应步骤大大简化。
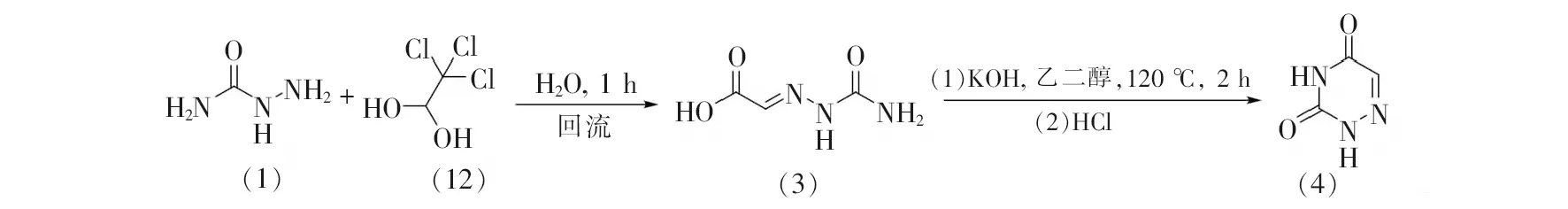
图3 6-氮杂脲嘧啶的改进合成方法
以上几种方法适用于6-氮杂脲嘧啶分子的合成,而针对含取代基的6-氮杂脲嘧啶类化合物,则需不同的方法合成。2016年,翟鑫等[13]以(2-氰基乙酰基)氨基甲酸乙酯(13)作为起始原料,与芳基重氮盐缩合生成腙类化合物(14),经过高温环化得到N1位芳基取代C5位氰基取代的6-氮杂脲嘧啶类化合物(15),收率62%~68%(图4)。该反应底物适用范围窄、官能团兼容性差,因而所得6-氮杂脲嘧啶种类单一。
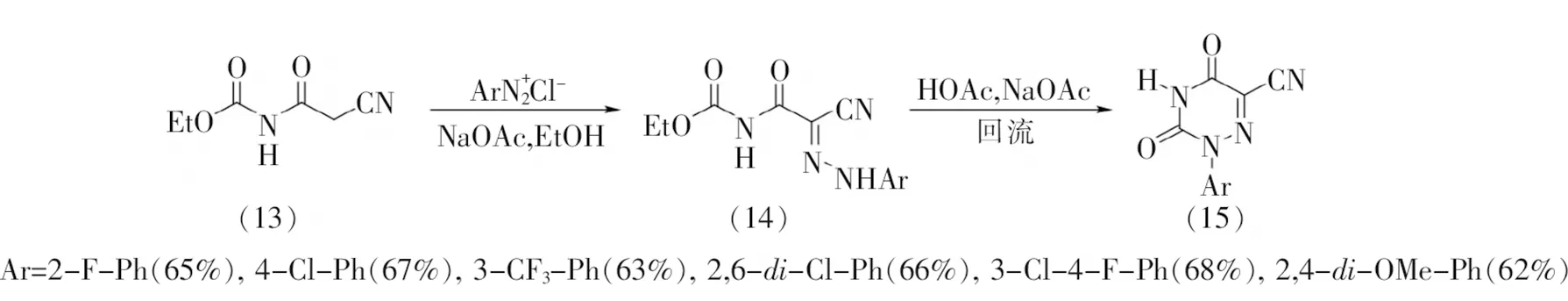
图4 1-芳基-5氰基-6-氮杂脲嘧啶类化合物的合成方法
2016年,Beauchemin等[14]以肼衍生物(16)为起始原料,原位生成异氰酸酯衍生物(17),进而与胺类化合物(18)发生亲核加成-环合串联反应合成了多种取代的6-氮杂脲嘧啶类化合物(20),收率35%~90%,见图5。该反应中伯胺、肼和羟胺均以中等到优异的产率生成目标产物,而且对于大位阻的环己胺、亲核性较低的苯胺以及含近端吸电子基的伯胺均能适应。此外,该串联反应对于不同取代基如甲基、苯基等均具有良好的耐受性,为多种6-氮杂脲嘧啶类化合物的合成提供了有效的方法。
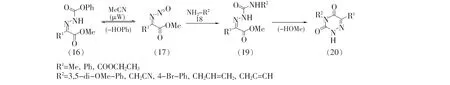
图5 具有取代基的6-氮杂脲嘧啶的合成方法
2 6-氮杂脲嘧啶的结构修饰方法
6-氮杂脲嘧啶类化合物构效关系的研究表明,可以通过修饰其结构获得具有不同生物活性的潜在药物分子。因此,探索对6-氮杂脲嘧啶结构的修饰方法对于新药的发现至关重要。
6-氮杂脲嘧啶分子氮原子的亲核性比较强,在碱性条件下易于与一级卤代烃如苄溴、烯丙基溴、溴代乙酸甲酯等发生亲核取代反应从而在氮原子上引入苄基、烯丙基等不同的取代基,并且通过调整伯卤代烃的用量可以选择性地生成N1位单取代的产物(22)或N1,3位双取代的产物(23)[15-16](见图6)。该法只适用于空间位阻较小的一级卤代烃,而对于二级卤代烷烃,则需要将6-氮杂脲嘧啶进行预处理活化才能进行反应。6-氮杂脲嘧啶先与N,O-双三甲硅基乙酰胺(BSA)或六甲基二硅氮烷(HMDS)反应生成活性中间体甲硅烷基化6-氮杂尿嘧啶(24),随后再与二级卤代烷烃或乙酰基化糖基作用生成N1位取代的6-氮杂脲嘧啶类化合物(27)[17-20]。通过此法可以合成多种重要的具有生物活性的6-氮杂尿嘧啶苷类化合物。
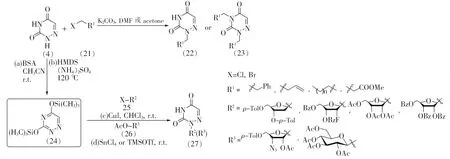
图6 基于亲核取代反应的6-氮杂脲嘧啶结构修饰方法
由于卤代苯sp2-碳卤键难于发生亲核取代反应,因而上述方法不适用于构建N-芳基取代6-氮杂脲嘧啶。杂原子芳基化则可通过铜催化的Chan-Lam偶联反应得以实现,此类反应条件比较温和,适用范围广,是构建碳杂键最有效和最直接的策略之一[21-23]。2018年,Seelam等[24]借鉴该策略以芳基硼酸(28)和6-氮杂脲嘧啶为起始原料,首次通过Chan-Lam偶联反应合成了18种N-芳基取代的6-氮杂脲嘧啶(29),收率60%~90%,见图7。
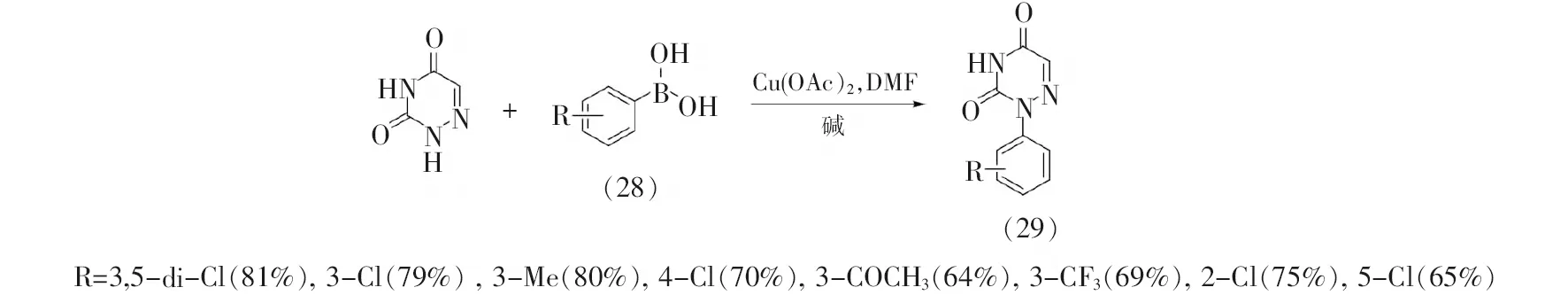
图7 基于Chan-Lam偶联反应的6-氮杂脲嘧啶结构修饰方法
上述亲核取代和交叉偶联反应适用于6-氮杂脲嘧啶环上的氮原子修饰,而由于碳氢键键能相对较高,6-氮杂脲嘧啶5号位碳原子修饰的研究进展则较为缓慢。起初,通过溴代反应预先在6-氮杂脲嘧啶的5号位碳原子引入溴原子,再以溴原子作为离去基团进行亲核取代反应或铃木偶联反应,从而引入不同取代基,如氨基、芳基和烷氧基等[4, 25-26](见图8)。但是该结构修饰方法步骤繁杂,不符合原子和步骤经济性原则。近年来,随着高原子经济性、高步骤经济性的碳氢键官能化策略的兴起,借鉴碳氢键官能化策略,直接在6-氮杂脲嘧啶5号位碳原子上引入官能团或取代基的方法逐渐建立起来,从而为高效地合成复杂、多样的C5位取代的6-氮杂脲嘧啶提供了新的途径[27-30]。
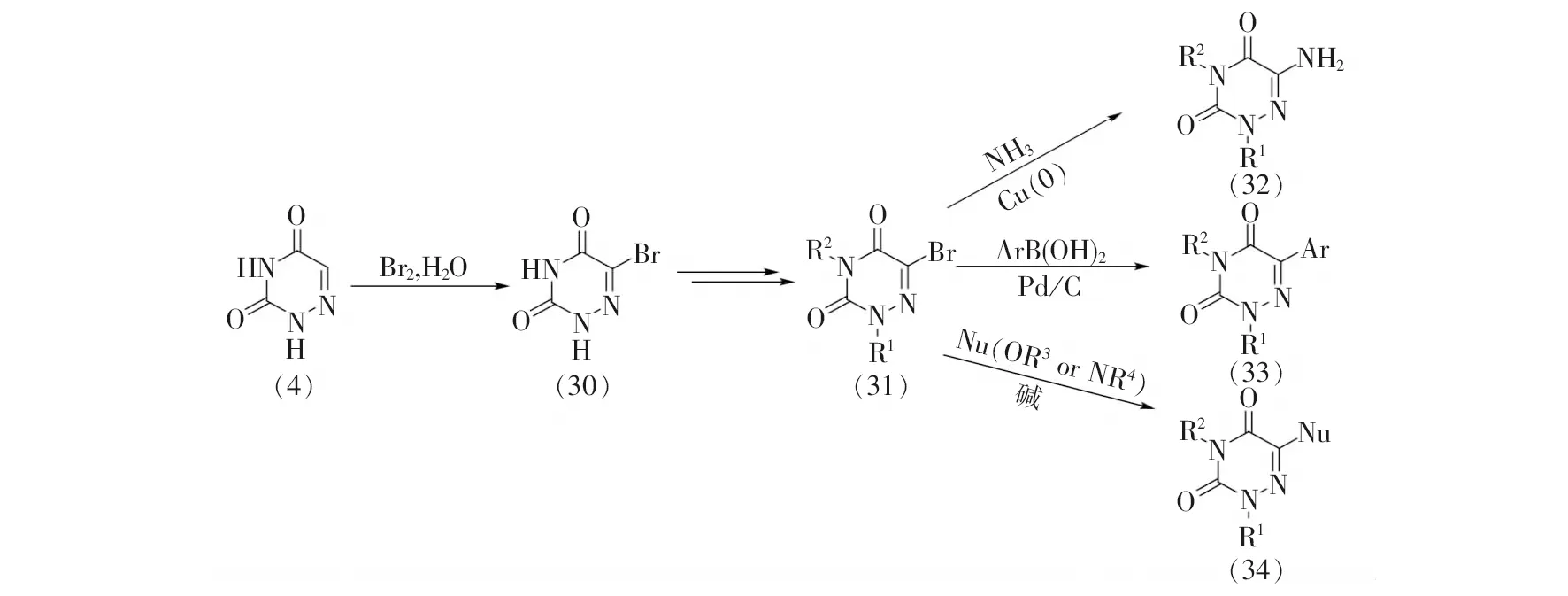
图8 基于溴化反应的6-氮杂脲嘧啶的结构修饰方法
6-氮杂脲嘧啶C5位碳氢键官能团化反应研究起步于2018年,6-氮杂脲嘧啶只是被视作底物拓展研究中的筛选对象之一,并未开展官能团兼容性等系统研究,且产率往往不高。如图9所示:上海有机化学研究所徐修华等[31]在研究杂环化合物与3,3,3-三氟-2,2-二甲基丙酸(35)的脱羧偶联反应中,将6-氮杂脲嘧啶作为候选底物之一,以42%的产率得到相应的目标分子(36);2020年南开大学李鑫等[32]在杂环化合物的二氟烷基化反应研究中,采用6-氮杂脲嘧啶作为底物、二氟甲基亚磺酸钠(37)作为二氟甲基化试剂,无需过渡金属催化剂和氧化剂,以玫瑰红(Rose Bengal)为光催化剂在可见光照射下产生的二氟甲基自由基引发反应,以73%的产率得到6-二氟甲基取代的6-氮杂脲嘧啶(38)。以上表明通过碳氢键官能化策略对6-氮杂脲嘧啶进行结构修饰的可行性。
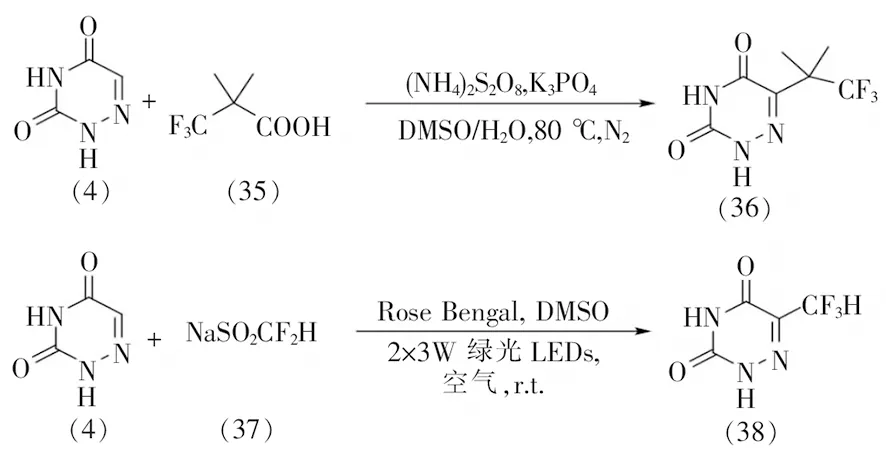
图9 基于碳氢键官能化策略6-氮杂脲嘧啶的结构修饰方法
2021年,Kim等[33]报道了6-氮杂脲嘧啶类化合物的碳氢键直接烷基化反应,见图10。
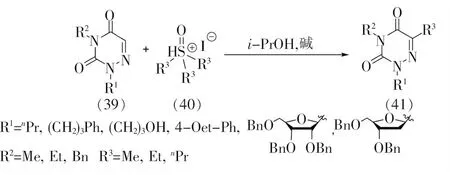
图10 6-氮杂脲嘧啶的烷基化
该反应以三烷基亚砜盐(40)作为烷基源,以中等到良好的产率获得C5位烷基化的6-氮杂脲嘧啶类化合物(41)。反应底物可以拓展到烷基、羟基和芳基等多种取代基N1位取代的6-氮杂脲嘧啶,展示了该反应在有机和药物合成中的应用潜力。
2021年,文献[34]报道了可见光诱导的6-氮杂脲嘧啶类化合物的碳氢键氨甲基化反应(见图11)。

图11 6-氮杂脲嘧啶的氨甲基化
该反应以N,N-二甲基苯胺为氮亚甲基源,在室温下反应生成15种C5位氨甲基取代的6-氮杂脲嘧啶(44),收率53%~80%。该反应还适用于其他N-甲基苯胺衍生物,如N,N-二乙基苯胺、各种N-烷基-N-甲基芳胺以及N-甲基苯胺等亲核试剂。此外,该反应还具有广泛的底物适用范围,甲基、苄基或烯丙基N1,3位取代的6-氮杂脲嘧啶均能以良好的产率得到相应氨甲基化产物,并且对苯环上无论是给电子还是吸电子基取代的N,N-二甲基苯胺均具有良好的耐受性。
2022年,Kim等[35]报道了钴(Ⅱ)催化的6-氮杂脲嘧啶类化合物的碳氢键烷基化反应,见图12。该反应以1,4-二氢吡啶衍生物(46)作为烷基化剂,实现了包括6-氮杂脲嘧啶在内的多种氮杂环的烷基化。该反应也适用于羟基、芳基和萘基等N-取代的6-氮杂脲嘧啶等底物,也可将异丙基引入N1位被脱氧核糖、核糖和木糖取代的6-氮杂脲嘧啶核苷类化合物(47)。
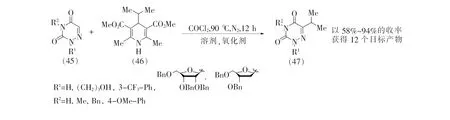
图12 6-氮杂脲嘧啶的烷基化
2022年,於兵等[36]在无过渡金属、无氧化剂条件下实现了自由基引发的6-氮杂脲嘧啶类化合物的碳氢键氰甲基化反应(见图13)。该反应以一种廉价的二硫代氨基甲酸盐阴离子为催化剂,实现了多种氮杂环的苄基化或氰甲基化。当以6-氮杂脲嘧啶作为底物时,以33%的产率获得氰甲基化的6-氮杂脲嘧啶(51)。该反应条件温和,为设计和合成新型药物和生物活性分子提供了一种有潜力的途径。
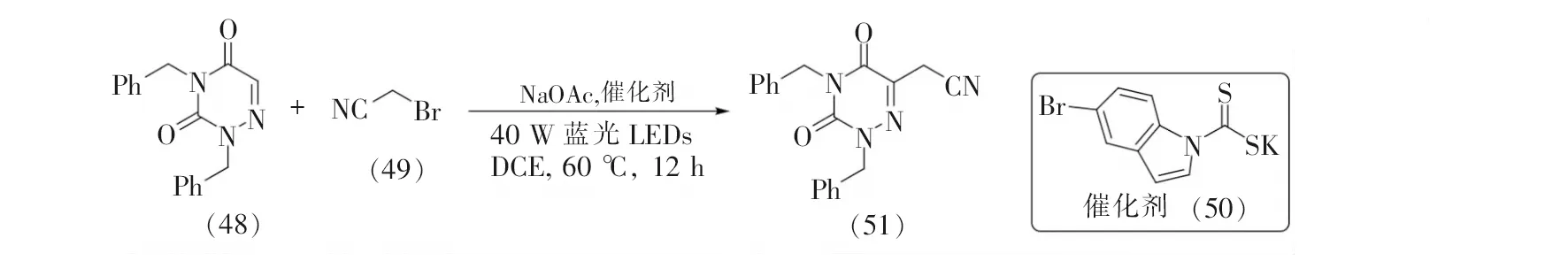
图13 6-氮杂脲嘧啶的氰甲基化反应
同年,於兵等[37]又报道了一种光照启动的自由基过程的6-氮杂脲嘧啶类化合物的碳氢键芳基化反应(见图14)。该反应无需光催化剂,而是芳基硫盐和三乙烯二胺(DABCO)原位生成的电子给体-受体(EDA)复合物在可见光诱导下产生自由基引发反应,最终成功地将芳基引入6-氮杂脲嘧啶骨架。该反应耐受甲基、甲氧基和氰基等多种基团,并且N3位没有取代基保护的6-氮杂脲嘧啶核苷类似物也顺利反应,以中等到良好的产率生成相应的芳基化产物(54)。
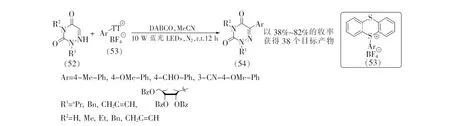
图14 6-氮杂脲嘧啶的芳基化
2022年,本课题组[38]报道了可见光照射下6-氮杂脲嘧啶类化合物的碳氢键氧烷基化反应(见图15)。
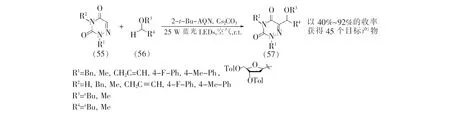
图15 6-氮杂脲嘧啶的烷氧基化
该反应中,多种N1,3位双取代的6-氮杂脲嘧啶以及N1位或N3位单取代的6-氮杂脲嘧啶都能在室温下顺利进行反应,以适中到良好的产率生成目标产物。从醚方面看,除了常用的环醚如1,4-二恶烷和四氢吡喃之外,开链的二丁醚和甲氧基环戊烷等也可作为烷氧基试剂,以良好的产率生成氧烷基化产物(57)。
3 结语与展望
6-氮杂脲嘧啶骨架是多种生物活性分子的构筑单元,研究6-氮杂脲嘧啶骨架及其修饰方法一直是有机及药物合成领域的内容之一。6-氮杂脲嘧啶的合成方法已很成熟,但存在反应路线长、原子经济性对环境不友好的问题,新的合成方法在一定程度上弥补上述缺点。此外,少数取代基取代的6-氮杂脲嘧啶骨架化合物,可以通过理性设计,缩短反应路线,提高合成方法的效率和原子经济型。目前,研究主要聚焦于6-氮杂脲嘧啶骨架N原子的烷基和芳基化,特别是C5位碳氢键直接官能化或修饰,初步发展了碳氢键的溴化、氟甲基化、氰甲基化、烷基化、芳基化、氨甲基化和烷氧基化新方法。
尽管在6-氮杂脲嘧啶类化合物的官能化或修饰方面取得了一些进展,但已获得的方法并非十全十美,普遍存在底物和官能团适应面较窄的问题。此外,一些反应需要过渡金属催化剂甚至为贵金属催化剂,而对于含6-氮杂脲嘧啶骨架药物合成而言,由于杂环结构与过渡金属的配位作用,产物与过渡金属难于分离,将存在金属残留问题。因此,未来研究方向将聚焦于底物与试剂适用面更加广泛的非过渡金属催化体系的构建,尤其是非过渡金属光催化剂的设计与反应的研究。
猜你喜欢
昆明医科大学学报(2021年8期)2021-08-13
武警医学(2018年10期)2018-11-06
石油石化绿色低碳(2018年5期)2018-03-20
石油炼制与化工(2016年6期)2016-04-06
当代化工研究(2016年6期)2016-03-20
合成化学(2015年2期)2016-01-17
合成化学(2015年2期)2016-01-17
化工进展(2015年6期)2015-11-13
中国塑料(2015年10期)2015-10-14
无机化学学报(2014年3期)2014-02-28
