
微波辅助下ZrO2/H-βMW催化剂的制备及其催化环己酮和异丙醇MPV反应研究
2023-11-22 02:34董林辉孟凡飞
精细石油化工 2023年6期
董林辉,孟凡飞,邱 俊*
(1.吉林化工学院化学与制药工程学院,吉林 吉林 132022;2.长春理工大学化学与环境工程学院,吉林 长春 130022)
MPV反应在有机合成中占有重要地位[1-5]。传统MPV反应常以金属醇盐作为催化剂,包括单金属醇盐、双金属醇盐、手性金属醇盐,分别如异丙醇铝、镨或钕等镧系双金属醇盐、钒的络合配体等[6-7]。这些均相催化剂的制备一般需要在无水无氧环境下醇与金属以加热回流的方法进行,操作过程复杂,对设备要求严格。MPV反应用到的多相催化剂主要有金属氧化物、沸石分子筛、负载型介孔分子筛和接枝金属醇盐的介孔分子筛[8-15]。近年来,在MPV反应的催化剂方面,关于分子筛的改性及均相催化剂的多相化的研究逐渐增多。
微波技术具有加热快速、均匀和渗透力强等特点,在沸石分子筛改性方面具有一定的优越性,越来越被受到关注和广泛应用[16-18]。据报道,微波辐射加热有利于活性组分分布均匀,提高了分散度,负载金属后分子筛的酸量和强度有明显提高,从而提高了催化性能。可见,借助微波辅助手段来开发制备新催化材料以实现有效的催化循环,提高其应用具有重大意义。有鉴于此,笔者在微波辅助下采用浸渍法制备了ZrO2/H-βMW催化剂,以环己酮和异丙醇的MPV还原为探针反应,评价了其催化性能,探究了催化剂的酸性、孔结构等与催化性能之间的关系。
1 实 验
1.1 主要试剂与仪器
环己酮,分析纯,天津市北辰方正试剂厂;异丙醇,分析纯,天津市永大化学试剂有限公司;八水合氧氯化锆,分析纯,国药集团化学试剂有限公司;H-β分子筛,南开大学催化剂厂;所有试剂使用前未经进一步提纯。
PANalytical X′Pert3粉末X射线衍射仪,荷兰马尔文帕纳科公司;AutoChem Ⅱ 2920全自动化学吸附仪,美国麦克仪器有限公司;Gemini VII 2390全自动比表面积测定仪,美国麦克仪器有限公司;ZeissSigma 300扫描电子显微镜,德国卡尔蔡司公司;Aglient 7800电感耦合等离子体质谱仪,美国安捷伦公司;STA449 F3同步热分析仪,德国耐驰公司。GC-MS QP2010气相色谱-质谱联用仪,日本岛津公司;GC-9720气相色谱仪,浙江福立仪器有限公司;MAS-Ⅱ微波合成仪,上海新仪微波化学科技有限公司。
1.2 催化剂的制备
按照氧化锆与H-β分子筛的质量比为2%、4%、6%、8%、10%的分布在微波反应仪中进行负载,将氧化锆换算成氧氯化锆的质量并配成100 mL溶液,按照固液比约为1∶10加入H-β分子筛约1.0 g,启动搅拌并设定微波功率加热至80 ℃后开始计时,10 min后反应结束,旋蒸去除水分,得白色固体粉末于烘箱中105 ℃干燥4 h。最后在马弗炉中程序升温焙烧(室温下以10 ℃/min速率升温至260 ℃,260 ℃温度下维持2 h,以15 ℃/min速率升温至550 ℃,在550 ℃焙烧6 h),研磨后得到的白色粉末分别记为2%ZrO2-H-βMW、4%ZrO2-H-βMW、6%ZrO2-H-βMW、8%ZrO2-H-βMW、10%ZrO2-H-βMW。
1.3 催化环己酮的MPV还原
典型的MPV反应如下:向微波反应仪内装有球形回流冷凝管的50 mL长颈三口烧瓶中依次加入0.79 g催化剂、12.12 g(0.201 mol)异丙醇和0.98 g(0.01 mol)环己酮,然后启动磁力搅拌并在400 W的微波功率下加热至80 ℃,反应15 min,冷却至室温,抽滤,催化剂滤饼于80 ℃干燥12 h。
1.4 产物分析
产物定性分析是在GC-MS QP2010仪器上进行,采用Rtx-5(30.0 m×0.32 mm×0.25 mm)毛细管色谱柱,主要操作条件为:N2作为载气,吹扫流量3.0 mL/min,进样口温度为250 ℃,检测器温度为290 ℃,进样方式为分流进样;程序升温起始温度为50 ℃,停留3 min,以10.00 ℃/min速率升温至150 ℃,停留5 min,最后以20.00 ℃/min速率升温至260 ℃,保持3 min。进样量为0.1 μL,电离方式为EI,电检测器温度280 ℃。
产物定量分析是在福立GC-9720上进行,采用Rtx-5(30.0 m×0.25 mm×0.3 mm)毛细管色谱柱,载气为N2,吹扫流量3.0 mL/min,FID检测器,进样口温度为250 ℃。检测器温度280 ℃。程序升温的起始温度为40 ℃,保留时间3 min;升温速率10.00 ℃/min升温到100 ℃,停留3 min;然后以30.00 ℃/min升到160 ℃,保留时间3 min;最后以30.00 ℃/min升到260 ℃,保留时间3 min。进样量0.1 μL。采用峰面积归一化方法,以反应物环己酮的量为基准,计算混合物中各组分含量,按下式计算环己酮转化率:

2 结果与讨论
2.1 催化表征
2.1.1 XRD
图1为H-β分子筛及微波辅助下其负载ZrO2后的粉末XRD谱。从图1可以看出,改性后的H-β分子筛与原分子筛的特征峰出现的位置一致,即在2θ为7.6°和22.4°处均出现特征衍射峰,说明这些催化剂均保持β分子筛的晶型和骨架结构。随着负载量的增加,特征峰出现宽化,强度减弱,结晶度(自JADE5.0软件)也逐渐减小。并且,试样并未观察到ZrO2的特征峰,表明ZrO2或以晶粒小于4 nm的微粒高度分散在分子筛的表面上,或Zr4+交换了骨架Al3+进入到了骨架中,而未被检测出。
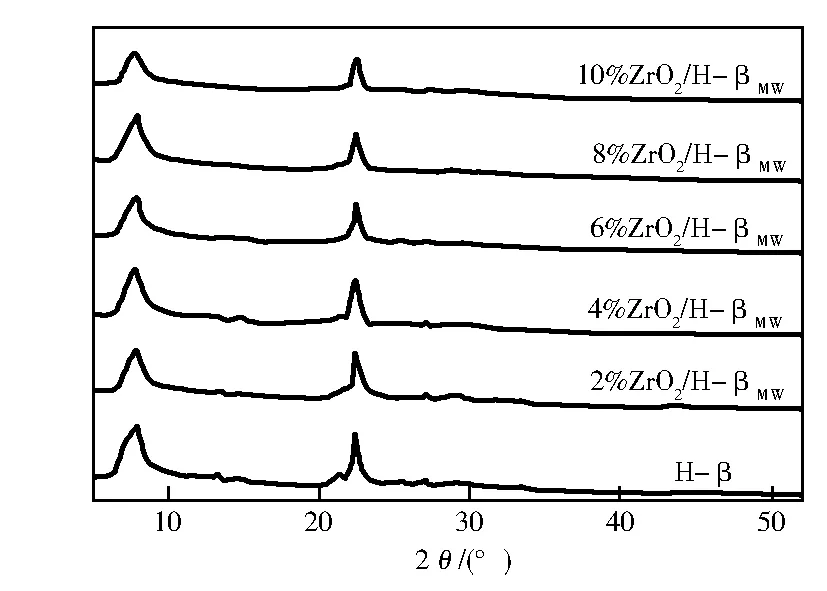
图1 试样的XRD谱
2.1.2 SEM
图2为H-β分子筛及其微波辅助下负载ZrO2后的SEM照片。从图2可以看出,H-β分子筛的形貌规整,棱角分明,而分子筛经过ZrO2改性后的晶粒虽然外形分明、结晶性良好,基本上分布均匀,但晶粒大小也略微出现差异,平均尺寸为200 nm。负载ZrO2后分子筛表面存在部分堆积晶粒,随负载量增加,极少部分堆积严重甚至覆盖分子筛晶粒导致集结,但没有杂晶和模糊不清的胶团,对H-β分子筛本身结构都没有太大的影响,这与XRD的表征结果也相一致。

图2 试样的SEM照片
2.1.3 NH3-TPD
ZrO2/H-βMW具有更多的表面酸性位,其Lewis酸中心会影响MPV反应的催化效率。图3为H-β分子筛及其微波辅助下负载ZrO2前后的NH3-TPD曲线。从图3可以看出,H-β分子筛及其微波辅助下负载不同量ZrO2后负载型分子筛的酸性强度及分布有一定的差别,它们的弱酸(160~170 ℃的峰)、中强酸(285~310 ℃的峰)中心都随ZrO2的负载量增加而向温度增加方向偏移,而且随着ZrO2负载量的增加,中强酸量略有增加,弱酸中心量也增加,总酸量增加,具体酸中心及其布数据见表1。结合表1各个催化剂的酸量数据及催化剂的催化环己酮和异丙醇MPV反应活性(表6),可以发现负载ZrO2后,弱酸量虽然增加但对催化MPV反应的活性没有明显影响,而中强酸量的变化对环己酮的MPV还原起到了决定性作用,中酸强度分布向强酸方向移动,有利于在催化剂表面产生催化作用而提高催化性能。

表1 NH3-TPD表征催化剂的酸量

图3 H-β改性后分子筛的NH3-TPD曲线
2.1.4 催化剂的BET比表面积和孔结构
图4为H-β分子筛及其微波辅助下负载ZrO2后的N2吸附脱附等温线,表2为H-β分子筛及其微波辅助下负载ZrO2后的比表面积、孔容和孔径数据。由图4可以看出,H-β分子筛及其微波辅助下负载ZrO2前后的吸附等温线均属于Ⅳ型等温线,表明这些催化剂均同时存在着微孔和介孔,而且它们的吸附量相差不很大,即随负载ZrO2的量增加其比表面积下降但不很明显。结合表2中数据可知,微波辅助下随着总孔容也呈现下降负载量的增加,负载型分子筛的BET比表面积呈现下降趋势,总孔容也呈现下降,而平均孔径出现先降低而随着ZrO2负载量的增加逐渐变大。因此,笔者认为ZrO2先是均匀高度分散使孔径变小而后随着ZrO2负载量的增加,发生更多的Zr4+和骨架Al3+的交换又使孔径逐渐变大;而随着ZrO2负载量的增加,无论高度分散还是骨架离子交换(Zr4+离子半径远大于Al3+离子半径),总孔容都会下降。虽然比表面积和孔容下降降低了载体H-β分子筛的催化活性,但由于负载ZrO2引入较多锆Lewis酸中心使得催化性能增加。

表2 H-β分子筛改性前后的BET比表面积、总孔容和平均孔径

图4 H-β分子筛负载ZrO2前后的N2吸附脱附等温线
2.1.5 ICP-MS
表3为H-β分子筛及其负载ZrO2后的ICP-MS数据。从表3可以看出,随着ZrO2负载量的增加,Si/Zr的摩尔比降低,这符合实验对氧化锆负载量增加的预期,但其实际测试量和理论预期值存在一定差别。Si/Al没有明显变化,与市售H-β分子筛的Si/Al相近。
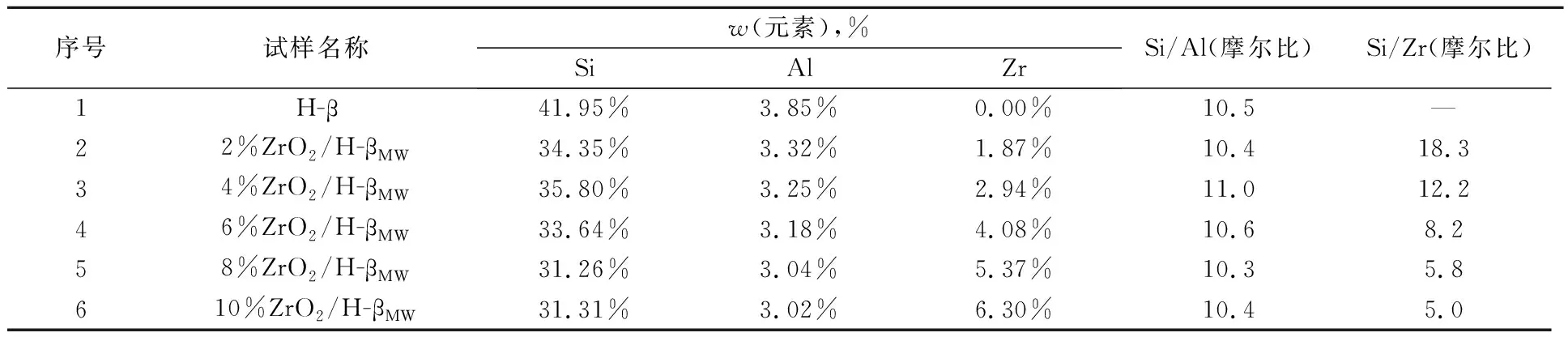
表3 H-β分子筛及其负载ZrO2改性后的ICP-MS数据
2.2 微波辅助工艺优化
2.2.1 催化剂制备因素考察
2.2.1.1 微波功率的影响
采用不同微波功率下制备的8%ZrO2/H-βMW催化剂,MPV反应的环己酮转化率见表4。由表4可知,适宜的微波功率400 W。

表4 微波功率对环己酮转化率的影响
2.2.1.2 微波下浸渍时间的影响
采用微波功率400 W条件下制备的8%ZrO2/H-βMW催化剂,不同微波浸渍时间下MPV反应的环己酮的转化率结果见表5。由表5可知,当催化剂的制备微波浸渍时间为10 min时,环己酮的转化率最高,因此后续实验采用微波条件下浸渍10 min制备催化剂。

表5 微波下浸渍时间对环己酮转化率的影响
2.2.1.3 ZrO2负载量的影响
固定微波功率400 W、浸渍时间10 min,考察ZrO2负载量对环己酮MPV反应转化率的影响,结果见表6。

表6 微波辅助下ZrO2负载量对环己酮转化率的影响
由表6可知,随着ZrO2负载量的增加,环己酮MPV反应的转化率也增加,当ZrO2负载量增加到10%时,环己酮的转化率达到96.3%。因此,适宜的ZrO2负载量为10%。
2.2.2 MPV反应因素考察
2.2.2.1 微波功率
以微波辅助优化条件下制备的10%ZrO2/H-βMW为催化剂,考察微波功率对环己酮转化率的影响,如表7所示。由表7可知,微波功率为400 W时环己酮转化率最高。

表7 微波功率对MPV反应环己酮转化率的影响
2.2.2.2 微波时间
以微波辅助最佳条件下制备的10%ZrO2/H-βMW为催化剂,微波功率为400 W,在回流温度下考察反应时间对MPV反应的影响,结果见表8。由表8可知,微波反应时间对MPV反应的影响不大,当微波反应时间为20 min时,环己酮的转化率较高。综合考虑,选定20 min为适宜反应时间。

表8 微波反应时间对环己酮转化率的影响
2.2.2.3 催化剂的用量
以微波辅助最佳条件下制备的10%ZrO2/H-βMW为催化剂,微波功率400 W,在典型的MPV反应条件下反应20 min,考察催化剂用量对MPV反应的影响,结果见表9。由表9可知,适宜的催化剂用量为6%。

表9 催化剂用量对环己酮转化率的影响
2.2.2.4 反应物配比
以微波辅助最佳条件下制备的10%ZrO2/H-βMW为催化剂,固定微波功率400 W、反应时间20 min、催化剂的用量6%,在典型反应条件下,考察反应物配比对MPV反应的影响,结果见表10。由表10可知,随着摩尔比的增加,环己酮的转化率逐渐增加,摩尔比为1∶20和1∶25时,环己酮的转化率相差不大。综合考虑,选定n(环己酮)∶n(异丙醇)=1∶20为适宜条件。

表10 反应物配比对环己酮转化率的影响
2.3 微波辅助工艺与传统条件下的比较
采用400 W微波辅助浸渍回流10 min得到的10%ZrO2/H-βMW催化剂,在上述优化MPV反应条件下,进行了3次平行实验,环己酮的平均转化率98.2%。说明微波辅助下制备的催化剂具有很好的催化活性稳定性,单因素实验优化后的实验工艺条件组合起来效果理想。另外,与传统加热工艺(反应时间5 h,环己酮转化率92.0%)比较,微波辅助工艺缩短了反应时间,并提高了转化率。
2.4 催化剂寿命
以10%ZrO2/H-βMW为催化剂,催化剂分别在焙烧和干燥后重复微波辅助下催化环己酮和异丙醇的MPV反应,循环使用5次,环己酮的转化率如表11所示。

表11 10%ZrO2/H-βMW催化环己酮和异丙醇MPV反应的循环使用数据
由表11可见,经过焙烧后重复使用的催化剂的活性远高于干燥后催化剂的活性:经过干燥后催化剂活性重复利用1次,其活性明显下降,随重复利用次数增加,活性急剧下降;而经过4次焙烧使用其活性略微下降,经5次焙烧再生后使用环己酮转化率有所下降至80.1%,说明焙烧能有效再生催化剂,除去催化剂表面及孔内使催化剂失活的水、二氧化碳和有机物等分子,而干燥只部分除去催化剂表面的物理吸附水、二氧化碳、有机物,不能很好地除去结合水、孔内或层间吸附水、气体分子和孔内有机物分子,致使造成催化剂失活。
图5为10%ZrO2/H-βMW催化剂经微波辅助催化环己酮和异丙醇MPV反应前后的TG-DSC曲线。从图5可以看出,反应前催化剂失重包括物理吸附水和孔内或层间吸附水,总失重约为9.3%;反应后催化剂失重分三个阶段,即:120 ℃以下的物理吸附水失重(6.3%),120~260 ℃的孔内或层间吸附水失重(3.2%),260~540 ℃有机物分子燃烧失重(5.6%),总失重约15.1%。根据反应后催化剂的DSC曲线,可知在300~600 ℃存在不可逆的氧化。

图5 10%ZrO2/H-βMW催化剂反应前后的TG-DSC曲线
3 结 论
a.微波辅助下,制备了ZrO2/H-βMW催化剂,其在催化环己酮和异丙醇MPV还原反应中表现出较高活性。适宜的制备条件为:在400 W的微波功率下浸渍10 min,ZrO2的负载量为10%;适宜的MPV还原反应条件为:微波功率400 W、n(环己酮)∶n(异丙醇)=1∶20、10%ZrO2/H-βMW用量6 %、反应时间20 min,环己酮转化率可达98.2%。
b.与传统方法(环己酮转化率92.0%)相比,微波辅助方法大大缩短反应时间并显著提高了环己酮的转化率,而且催化剂可回收再利用,有效节约了资源和降低能耗。
c.负载型分子筛催化剂ZrO2/H-βMW比表面积和总孔容随着ZrO2的载入呈现下降趋势,平均孔径无明显变化规律,ZrO2的载入使得Si/Zr原子比下降,由于引入锆Lewis酸中心使中强酸分布移向强酸方向,导致极大提高了环己酮的转化率。
d.催化剂经TG-DSC热分析表明,反应前失重主要来自吸附的水分脱除,反应后催化剂孔道内有少量有机物滞留,经焙烧再生后被除去使催化剂恢复活性。
猜你喜欢
化学工程师(2023年1期)2023-02-17
云南化工(2021年7期)2021-12-21
有机氟工业(2021年3期)2021-09-15
化工管理(2021年7期)2021-05-13
理化检验-化学分册(2020年12期)2021-01-26
中国果树(2020年2期)2020-07-25
上海农业科技(2019年1期)2019-02-22
潍坊学院学报(2016年6期)2016-04-18
石油炼制与化工(2016年4期)2016-04-06
合成化学(2015年10期)2016-01-17
