
纳米薄片组装的花状NiMoS催化剂制备及其加氢脱硫性能
2023-11-16 08:53袁胜华李唯楚魏春金郑进保方维平伊晓东赖伟坤
石油学报(石油加工) 2023年6期
袁胜华,李唯楚,魏春金,郑进保,方维平,伊晓东,赖伟坤
(1.中石化(大连)石油化工研究院有限公司,辽宁 大连 116041;2.厦门大学 化学化工学院 醇醚酯化工清洁生产国家工程实验室,福建 厦门 361005;3.集美大学 理学院,福建 厦门 361021)
过渡金属硫化物具有独特的晶体结构和化学性能,被广泛应用于多相催化[1]、锂离子电池(LIBs)[2]、电化学电容器[3]及潜在的氢燃料电池[4-5]等。作为最重要的催化材料之一,基于Ni/Co促进的硫化钼或硫化钨催化剂已被应用于石油精制加氢脱硫(HDS)等[6-7]。传统氧化铝负载的Ni(Co)-Mo(W)硫化态HDS催化剂通常由其相应的金属氧化物经过高温硫化制得[8],但这些催化剂的性能仍然难以满足深度HDS的要求,其催化活性受限于催化剂活性位点的数目和活性位结构。非负载型硫化态催化剂由于具有硫化完全及单位面积上活性位点数目多等特征,表现出很高的HDS反应活性,因而成为目前HDS催化剂研究热点之一。近年来,有研究者以四硫代钼酸铵(ATTM)、镍前驱物及氢气为原料采用水热法或热分解法合成了非负载型硫化钼催化剂,相比于商业的Ni(Co)MoS/Al2O3催化剂,该方法合成的催化剂表现出优异的HDS活性[9-11]。
考虑到MoS2催化剂活性位与其形貌及尺寸密切相关,多种纳米结构MoS2已被成功合成[12-13],如纳米片、纳米薄片等[14-15]。目前已有较多文献报道采用多种方法合成了MoS2纳米花[16-17],所合成纳米花由大量纳米片状花瓣组成,纳米花球直径一般为数百纳米,每个花瓣厚度一般为十余纳米。这一特定纳米花状结构具有表面积大及形貌可控等特点[18-20],其在催化材料领域具有较好的应用前景。然而,无Ni/Co促进的MoS2纳米材料的HDS性能显著受限于其较差的活性位结构。一般地,MoS2与Co或Ni结合能够明显促进其对HDS反应的催化效果。因此,MoS2纳米材料在HDS中的应用重点在于保持其纳米结构的同时引入Ni/Co组分。不同形貌的纳米催化剂的HDS活性成倍高于可比较的商业催化剂,且其烯烃加氢活性也明显较高[21-22]。但是,目前关于Ni/Co促进的MoS2特定纳米结构催化剂的报道仍然较少。
综上,NiMoS催化剂被广泛应用于石油加工的HDS过程,作为潜在的HDS催化剂,NiMoS纳米材料的发展受到密切关注[23-24]。笔者系统研究了花状NiMoS纳米催化剂的合成及其催化HDS活性,以水合肼作为还原剂、单质硫作为硫源,采用水热硫化法成功合成了由尺寸均一的纳米薄片组成的花状NiMoS催化剂,采用XRD、SEM和TEM等手段对花状NiMoS催化剂进行表征,考察了其应用于噻吩和4,6-二甲基二苯并噻吩(4,6-DMDBT)的HDS催化反应性能。
1 实验部分
1.1 原料和试剂
硝酸镍(Ni(NO3)2·6H2O,质量分数98.5%)、钼酸铵((NH4)6Mo7O24·4H2O,质量分数99.9%)、水合肼(N2H4·H2O,质量分数85%)、单质硫(S,质量分数99.5%),硫化铵((NH4)2S,质量分数99.5%),均为分析纯,国药集团化学试剂有限公司产品;噻吩(C4H4S,质量分数99%)、4,6-DMDBT(C14H12S,质量分数97%),均为分析纯,Alfa Aesar化学有限公司产品;商用氧化铝载体(Al2O3),比表面积226 m2/g,由中石化(大连)石油化工研究院有限公司提供。
1.2 催化剂制备
以水合肼作为还原剂、单质硫作为硫化剂,采用水热硫化法探索合成由纳米薄片组装的三维花状NiMoS催化剂,合成过程如图1所示。将1.000 g (NH4)6Mo7O24·4H2O和0.844 g Ni(NO3)2·6H2O溶解于10 mL的去离子水中,超声振荡30 min后加入一定量的单质硫分散均匀,然后加入8 mL的水合肼溶液充分混合;将混合溶液转移至25 mL的水热合成釜中,置于180 ℃的烘箱中进行水热处理4 d;水热完成后在室温下自然冷却,对黑色沉淀物进行离心分离,用去离子水、稀盐酸和乙醇各洗涤数次,最终在80 ℃下真空干燥24 h得到催化剂样品。分别按单质硫加入质量0.25、0.50和0.75 g计算其理论质量分数分别为50%、100%和150%,所制备的NiMoS催化剂分别标记为NiMoS-50%S、NiMoS-100%S和NiMoS-150%S。单质硫质量分数100%、不同水热温度(150、180、200 ℃)处理4 d制备的NiMoS催化剂分别标记为NiMoS-100%S-150℃、NiMoS-100%S-180℃和NiMoS-100%S-200℃。单质硫质量分数100%、在水热温度180 ℃下处理不同时间(1、2、3、4、7 d)制备的NiMoS催化剂分别标记为NiMoS-100%S-1d、NiMoS-100%S-2d、NiMoS-100%S-3d、NiMoS-100%S-4d和NiMoS-100%S-7d。

图1 纳米薄片组装的花状NiMoS催化剂的水热硫化合成示意图
参比催化剂的制备:按上述NiMoS催化剂的合成过程,用(NH4)2S溶液替代水合肼还原剂和单质硫硫化剂,制备片状NiMoS催化剂,标记为NiMoS-F。按照文献[25]报道方法合成体相NiMoS催化剂,将Ni(NO3)2·6H2O和(NH4)6Mo7O24·4H2O混合溶液在80 ℃下缓慢蒸发去除水分,再在110 ℃下干燥,500 ℃空气气氛下焙烧5 h,最后在0.1 MPa、H2S/H2(H2S体积分数15%)气氛下400 ℃硫化2 h制得体相NiMoS催化剂,标记为NiMoS-B。按照文献[26-27]所述共浸渍法制备NiMo/Al2O3催化剂,将一定量的Ni(NO3)2·6H2O和(NH4)6Mo7O24·4H2O配制成混合浸渍液浸渍载体Al2O3,110 ℃干燥16 h,再在500 ℃焙烧5 h,最后在0.1 MPa、H2S/H2(H2S体积分数15%)气氛下400 ℃硫化2 h制得硫化态NiMo/Al2O3催化剂。所有参比催化剂的Ni/Mo原子比均为0.5。
1.3 催化剂表征
采用Rigaku公司生产的Ultima Ⅳ型X射线粉末衍射仪表征催化剂样品的晶体结构,条件为:管电流35 mA,管电压40 kV,X′Celerator超能阵列探测器,以CuKα(λ=0.15406 nm)为辐射源,测试范围(2θ)为10°~70°,采用normal扫描方式进行测试。所得谱图均经过X′pert Highscore软件处理,进行解谱分析,并用JCPDS文件数据库进行峰鉴定。采用Zeiss公司生产的SIGMA型场发射扫描电子显微镜观察催化剂样品的微观形貌,发射电压10 eV,加速电压为20 kV。采用Philips公司生产的FEI Tecnai 30型透射电子显微镜观测催化剂上MoS2的结构与分布,仪器电压300 kV。测试催化剂样品研磨粉碎后,加入到无水乙醇中超声波分散30 min,然后取少量悬浮液滴到表面附有碳膜的铜栅格子中,晾干,然后进行观测分析。
1.4 HDS活性测试
NiMoS催化剂上噻吩的HDS活性评价实验在固定床常压微型反应器上进行。反应器中反应管为内径5 mm石英管,催化剂装填量为200 mg,颗粒粒径40~60目。催化剂首先在H2气氛下(体积流速40 mL/min)以升温速率5 ℃/min升至300 ℃,常压预处理2.0 h。预处理完成后,反应器降温至所需反应温度250 ℃,由纯H2将恒温鼓泡器中的噻吩蒸汽带入反应器,通过控制噻吩蒸汽压和H2压力实现噻吩/H2分压比的调节。产物的在线分析在上海伍豪GC-9560气相色谱仪上进行,配有SE-30毛细管色谱柱和氢焰检测器(FID)以及一套全自动六通定量阀。NiMoS催化剂上噻吩的HDS活性以噻吩的HDS转化率(X,%)为评价指标,计算如式(1)所示。
(1)
式中:Cbutane和Cthiophene分别为反应器出口检测到的丁烷和噻吩的浓度,mol/L。
4,6-DMDBT的HDS活性评价在固定床连续流动高压微型反应器上进行,采用十氢萘溶解质量分数0.20%的4,6-DMDBT作为模型反应物[28]。催化剂装填量为500 mg,催化剂颗粒过筛至40~60目,且用石英砂稀释保持总体积固定为1.0 mL。反应前催化剂首先在40 mL/min的H2气流下,以升温速率5 ℃/min升温至300 ℃进行常压预处理2.0 h。HDS反应条件为:温度350 ℃,H2压力3.0 MPa,H2与液体原料的体积比600,液相体积空速4.0 h-1。反应稳定2.0 h后每隔1.0 h进行1次采样分析,共进行6次采样,油品硫含量分析在配有HP-5毛细管色谱柱和火焰光度检测器(FPD)的GC-9560气相色谱仪上进行,取6次分析结果的平均值作为最终的硫含量。4,6-DMDBT的HDS活性以反应产物中的硫质量分数(μg/g)为评价指标。
2 结果与讨论
2.1 NiMoS催化剂制备条件的优化
NiMoS催化剂制备过程的优化主要考察了硫化剂、水热硫化温度和时间的影响,并采用XRD、XPS、SEM和TEM等手段对不同制备条件下的NiMoS催化剂样品进行表征,同时考察催化剂在温度为250 ℃、H2压力为0.1 MPa、n(H2)/n(Thiophene)为10、噻吩空速为1.4×10-5mol/(g·s)的条件下噻吩的HDS转化率。
2.1.1 硫化剂的影响
NiMoS催化剂的制备过程中以单质硫作为硫化剂,考察了单质硫不同用量制备的NiMoS催化剂样品的XRD谱图、XPS谱图及对噻吩HDS转化率的影响,结果见图2。由图2(a)可知,NiMoS-50%S催化剂除了出现归属于立方晶系的NiS2(JCPDS no.89-3058)和斜六方晶系的NiS (JCPDS no.86-2280)衍射峰,还存在未硫化的立方晶系Mo3O (JCPDS no.72-0527)的衍射峰,说明当硫化剂不足时,镍的硫化优先于钼并消耗了大部分硫,因而钼未能完全硫化;NiMoS-100%S催化剂可明显观察到MoS2和NiS2的衍射峰,表明硫化反应进行较为完全;NiMoS-150%S催化剂MoS2的(002)晶面的衍射峰有明显的宽化现象,结晶度有所下降。由图2(b)可知,NiMoS-150%S催化剂中有单质硫残余。由图2(c)可知,NiMoS-100%S催化剂的HDS活性明显较高;结合XRD和XPS S 2p谱图分析可知,NiMoS-50%S催化剂中Mo未能完全硫化,因而其HDS活性较差;NiMoS-150%S催化剂存在未反应的单质硫杂质,导致催化剂的HDS活性明显降低。
2.1.2 水热硫化温度的影响
图3为不同水热温度下制备的NiMoS-100%S催化剂样品的XRD和XPS谱图。由图3(a)可知:NiMoS-100%S-150℃催化剂晶型较差,虽能观察到MoS2和NiS2的衍射峰,但同时也存在一些中间产物如Ni(OH)2(2θ=11.3°);NiMoS-100%S-160℃催化剂中间产物基本消失,但产物结晶度仍然较低;NiMoS-100%S-180℃催化剂MoS2的(002)晶面的衍射峰有明显的宽化现象,且NiS2的结晶度也有所提高;而NiMoS-100%S-200℃催化剂MoS2的(002)、(100)和(103)晶面衍射峰明显增强,且NiS2的衍射峰也显著提高,说明MoS2和NiS2的晶粒尺寸增大。在XRD谱图中,随着水热温度的升高,MoS2和NiS2的衍射峰逐渐增强,MoS2片层结构形成且叠加变厚。XPS谱图显示,相比于NiMoS-100%S-180℃,NiMoS-100%S-200℃样品的硫化度(Mo4+占比)从91%提高至95%,且NiMoS比例也从75%增大到77%,如图3(b)和图3(c)所示。

图3 不同水热温度下制备的NiMoS-100%S催化剂样品的XRD和XPS谱图
图4为不同水热温度所制得的NiMoS-100%S样品的SEM和TEM照片。由图4可知:NiMoS-100%S-150 ℃样品为少量的薄片杂乱堆积而成,但仍有大部分产物未成形且呈无规则颗粒状;NiMoS-100%S-180℃样品基本上都呈现花球状结构,花球尺寸均一且直径介于300~600 nm,花球上花瓣由超薄纳米片组装而成;NiMoS-100%S-200℃样品可以观察到明显的花球直径增大且组成片层厚度提高,MoS2薄层沿垂直于c轴的方向延伸,并存在自身的卷曲变形。同时,水热温度升高后,样品的结晶度升高,MoS2平均晶条长度由10.6 nm增大至21.0 nm,平均堆垛层数由5.8提高至7.8。因此,在水热硫化条件下,随着水热温度的升高,水热压力也不断增加,产物在温度和压力作用下晶粒不断长大、重排,并沿着c轴方向逐渐叠加和堆积[29],最终形成厚度为6~12 nm的纳米薄片,并组装成纳米花球。
对不同水热温度制得的NiMoS催化剂进行噻吩HDS活性测试,结果如图5所示。由图5可知,NiMoS-100%S-180℃催化剂的HDS活性明显较高,结合XRD和SEM分析结果可知,NiMoS-100%S-150℃样品结晶度较低,硫化未完全,且花球状结构亦未完全成形,因而HDS活性较差。而NiMoS-100%S-200℃样品MoS2晶条长度和堆垛层数明显增大、片层厚度提高,Mo分散度由0.135下降至0.082,导致边角S空位数目明显减少,边角S空位的数量是决定HDS活性的主要因素,因而所制得催化剂的HDS活性显著降低。
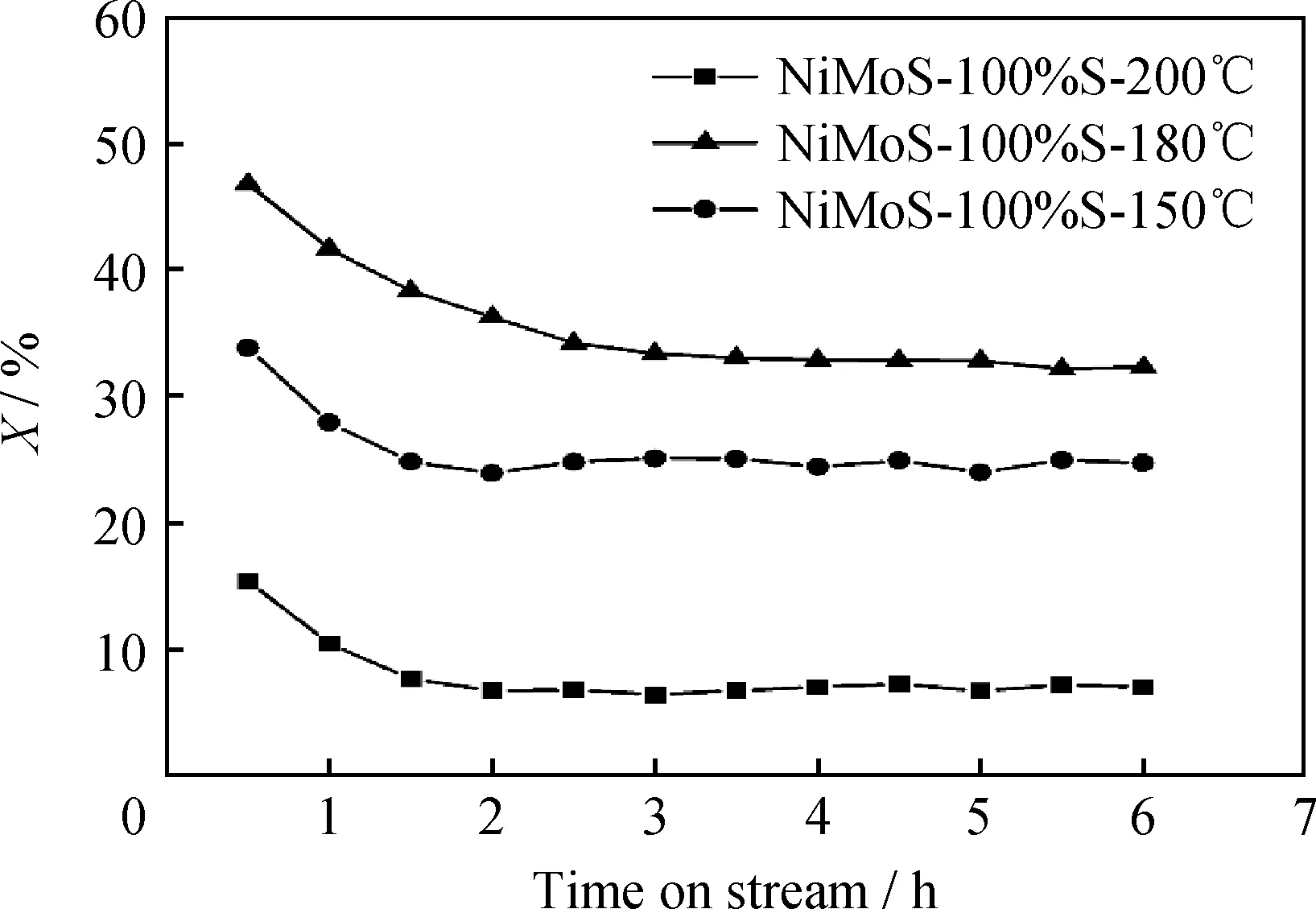
图5 不同水热温度下制备的NiMoS-100%S催化剂对噻吩HDS转化率(X)的影响
2.1.3 水热硫化时间的影响
为了研究NiMoS-100%S催化剂水热合成各阶段的硫化情况以及形貌结构变化,对不同水热时间下制备的NiMoS-100%S催化剂样品进行XRD和XPS表征,结果见图6。由图6(a)可知:NiMoS-100%S-1d样品Mo的硫化反应基本未进行而Ni的硫化也不完全,因而只能观察到较弱的NiS2衍射峰;NiMoS-100%S-2d样品NiS2结晶度明显升高,Ni硫化较为完全,但MoS2的衍射峰仍基本未见;NiMoS-100%S-3d和NiMoS-100%S-4d样品NiS2基本没有变化,而MoS2(002)晶面的衍射峰逐渐增强,说明Mo硫化反应的进行及MoS2层状结构逐渐形成;而NiMoS-100%S-7d样品MoS2的(002)、(100)和(103)晶面衍射峰均明显增强且有宽化现象,说明Mo的硫化反应进行较为完全,结晶度提高。由图6(b)和(c)可知,与NiMoS-100%S-3d催化剂相比,NiMoS-100%S-4d催化剂中Mo4+占比从77%提高至91%,NiMoS相占比从65%提高至75%,说明NiMoS-100%S-4d催化剂的硫化程度增加。
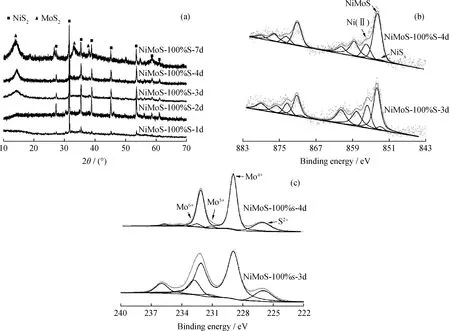
图6 不同水热时间制备的NiMoS-100%S催化剂样品的XRD和XPS谱图
图7为不同水热时间所制得的NiMoS-100%S样品的SEM照片。由图7可知:NiMoS-100%S-1d样品呈无规则颗粒状或者块状杂乱堆积,其表面未有纳米片的生成;NiMoS-100%S-2d样品仍是呈无规则颗粒状或块状,但有部分团簇表面已有少量纳米片生成;NiMoS-100%S-3d和NiMoS-100%S-4d样品基本上都呈花球状结构且花球尺寸均一,花球上花瓣由大量超薄纳米片组装而成;而NiMoS-100%S-7d样品可以观察到花球直径略有增大,且其组装的纳米薄片厚度也略有升高。结合XRD分析可知,在水热反应初期,Ni的硫化优先于Mo进行,片层状MoS2仍未成形,产物呈无规则颗粒状或块状;而在水热反应中后期,MoS2的晶面衍射峰逐渐增强,MoS2晶片生长以及花球状三维结构进行组装。

图7 不同水热时间制备的NiMoS-100%S催化剂样品的SEM照片
对不同水热时间所制得NiMoS-100%S催化剂进行噻吩HDS活性测试,结果见图8。由图8可知,相比于NiMoS-100%S-1d,NiMoS-100%S-2d催化剂的噻吩HDS活性显著提高,这是因为NiMoS-100%S-1d催化剂中Mo的硫化度较低,因而HDS活性较差。NiMoS-100%S-3d和NiMoS-100%S-4d催化剂MoS2晶片逐渐生长,且花球状形貌结构逐渐组装成形。NiMoS-100%S-4d催化剂的硫化程度增加,NiMoS活性位逐渐增多,因而其HDS活性逐渐升高。而NiMoS-100%S-7d催化剂的HDS活性略有下降,说明水热时间过长时,虽然其形貌结构变化不明显,但样品的结晶度提高,MoS2晶条长度有所增加。
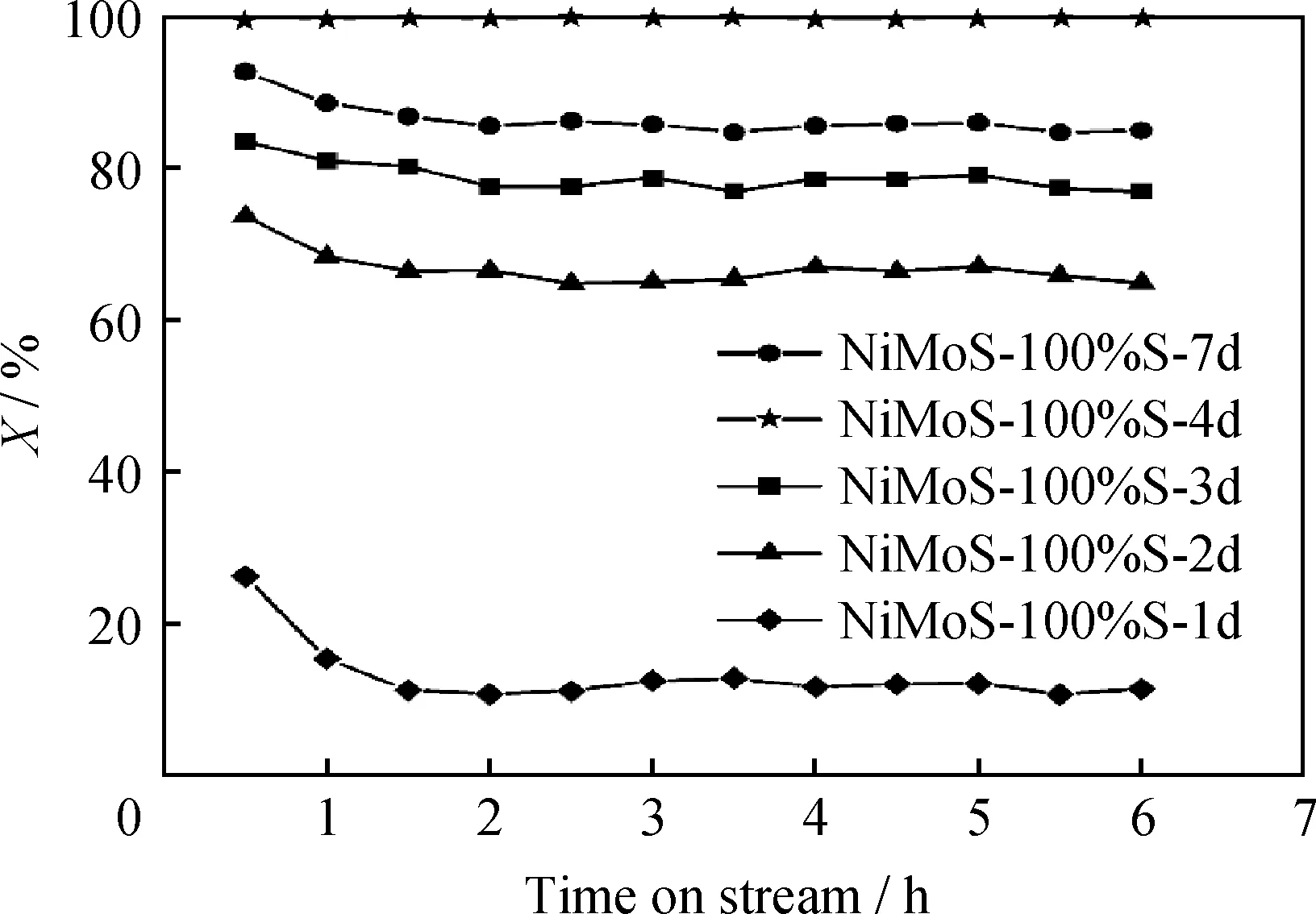
图8 不同水热时间下制备的NiMoS-100%S催化剂对噻吩HDS转化率(X)的影响
2.2 NiMoS催化剂的反应活性与稳定性
在上述制备过程优化的基础上,进一步考察了最佳合成条件下制备的NiMoS-100%S催化剂及片状NiMoS-F、体相NiMoS-B、NiMoS-A、负载型NiMo/Al2O34种参比催化剂对噻吩HDS转化率和4,6-DMDBT的HDS反应后硫含量的影响,结果见图9。由图9可知:纳米花状NiMoS-100%S和纳米片状NiMoS-F催化剂的噻吩HDS转化率明显高于体相NiMoS-B及NiMo/Al2O3催化剂;纳米花状NiMoS-100%S催化剂反应后油品中的硫残余质量分数仅为8.5 μg/g,其脱硫率远高于片状NiMoS-F和体相NiMoS-B催化剂,说明纳米花状NiMoS-100%S能够高效催化转化4,6-DMDBT。以四硫代钼酸铵为前驱物制备的NiMoS-A和负载型NiMo/Al2O3催化剂具有较好的脱硫活性,反应后油品中硫残余质量分数分别为18和30 μg/g。在NiMo含量一致的情况下,笔者制备的纳米花状NiMoS仍然具有相对较高的脱硫活性。由于NiMoS催化剂表面具有大量较短的MoS2晶条,拥有更多的S空位活性位点,因而其具有优异的HDS活性。
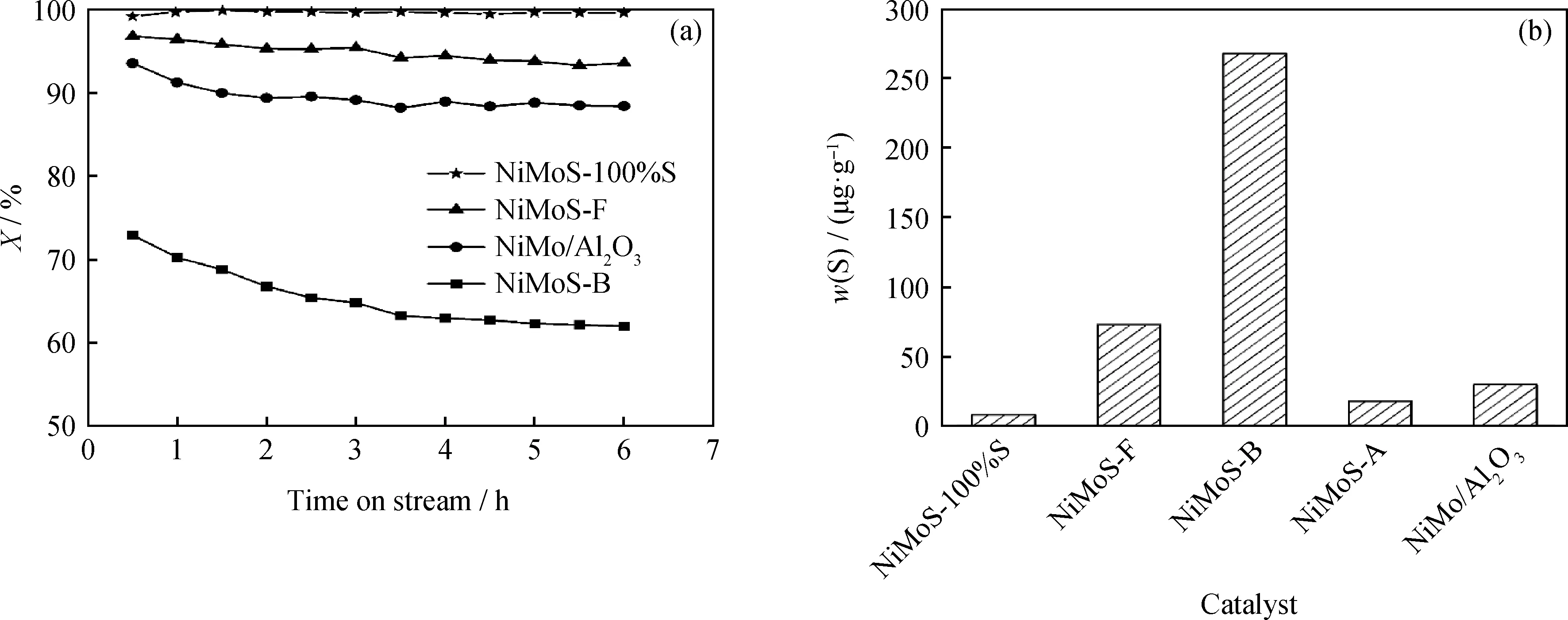
图9 NiMoS-100%S催化剂及4种参比催化剂对噻吩HDS转化率(X)和4,6-DMDBT的HDS反应后硫含量的影响
NiMoS-100%S新鲜剂经H2还原活化后进行噻吩HDS反应,过程中催化剂的XRD谱图结果如图10所示。由图10可知,NiMoS-100%S新鲜剂经过300 ℃、H2气氛还原后,其MoS2对应的衍射峰明显增强,结晶度提高;而NiS2晶相被还原为Ni3S2,结晶度有所下降。说明部分Ni再分散,与MoS2作用生成NiMoS。经过噻吩HDS反应后,MoS2对应的衍射峰又有所减弱,结晶度降低;而Ni3S2晶相转变为NiS。说明硫化镍晶型随反应气氛变化而发生改变。

图10 NiMoS-100%S催化剂的新鲜剂、还原剂及反应剂的XRD谱图
为了对比NiMoS-100%S催化剂微观形貌的稳定性,对还原剂和反应剂进行SEM和TEM表征,结果如图11所示。由图11可知,NiMoS-100%S催化剂进行H2还原活化处理后,其花球状结构未发生明显改变,花球上的超薄纳米片界限清晰且厚度相当,但花球之间堆积更加紧密,纳米薄片MoS2硫化完全且分散性较好。当NiMoS-100%S催化剂进行噻吩HDS反应后,可以明显地观察到花球接合在一起形成更大的花簇结构,但其纳米薄片结构仍然存在且厚度也未有明显变化。TEM照片统计分析结果显示,纳米薄片上MoS2晶条长度由10.6 nm增大至18.8 nm。综上可知,NiMoS-100%S催化剂经300 ℃、H2气氛活化及HDS反应后,其花球结构堆积结合更加紧密,而纳米薄片结构基本能够维持,不因高温及H2气氛作用使晶粒不断长大而破坏其微观结构。
3 结 论
(1)以单质硫作为硫化剂,水合肼作为还原剂,采用水热硫化法成功合成了由纳米薄片组装的NiMoS三维纳米花结构催化剂。在合成过程中,单质硫含量、水热反应时间和水热温度对NiMoS纳米花状结构有着重要影响。单质硫添加质量分数100%、在180 ℃温度下水热反应4 d时所制得催化剂中Mo和Ni前驱物硫化完全,呈三维花球状结构,花球尺寸均一且直径介于300~600 nm,花球上花瓣由厚度为6~12 nm的纳米薄片组装而成,纳米薄片上MoS2晶条可暴露更多的边角S空位。
(2)与其他4种参比催化剂片状NiMoS-F、体相NiMoS-B、NiMoS-A、负载型NiMo/Al2O3相比,180 ℃水热反应4 d所制备的NiMoS-100%S催化剂具有更优异的HDS催化反应活性。在温度为250 ℃、H2压力为0.1 MPa、n(H2)/n(噻吩)为10、噻吩空速为1.4×10-5mol/(g·s)的条件下,NiMoS-100%S催化剂作用下噻吩HDS转化率高于99.6%。结合XRD、SEM、TEM的分析结果表明,NiMoS-100%S催化剂的纳米花球的纳米薄片结构能够在高温还原及HDS反应过程中保持稳定。纳米薄片组装的花状NiMoS复合材料对于油品HDS过程具有潜在的应用价值。
猜你喜欢
科普童话·学霸日记(2021年4期)2021-09-05
装备制造技术(2020年4期)2020-12-25
学生天地(2017年30期)2018-01-05
学生天地·小学低年级版(2017年10期)2017-12-11
当代化工研究(2016年1期)2016-03-16
无机盐工业(2016年4期)2016-03-15
合成化学(2015年10期)2016-01-17
应用化工(2014年9期)2014-08-10
应用化工(2014年9期)2014-08-10
无机化学学报(2014年12期)2014-02-28
