
基于分子模拟的石墨烯表面含氧官能团对氢气吸附特性
2023-11-16 08:52刘亚楠陈叔平史庆智刘竞中赵碧婧
石油学报(石油加工) 2023年6期
刘亚楠,陈叔平,史庆智,刘竞中,赵碧婧
(兰州理工大学 石油化工学院,甘肃 兰州 730050)
在低温容器常采用的高真空多层绝热结构中,夹层真空度为0.01 Pa时,才能展现出其最佳的绝热效果。由于低温容器的漏气和夹层材料的放气,其真空度逐渐遭到破坏,缩短低温容器的全周期寿命[1-2]。氢气是引起低温储罐高真空绝热夹层真空失效的主要气体成分[3-5],在真空夹层中放置适当的吸气剂成为延长夹层真空寿命的有效方法。
无机碳材料,如活性炭、碳纳米管、石墨烯等,因具有比表面积高、稳定性好等优点,被广泛应用于吸附领域[6-9]。自2004年Novoselov等[10]采用撕胶带法将石墨烯从石墨中分离,这种二维材料便引起了人们极大的关注。很多学者研究石墨烯的吸氢性能,发现其在低温高压下对氢气有一定的吸附能力[11-14],Eftekhari等[15]指出理想的扁平石墨烯片虽可提供较高的比表面积,但六角形排列的sp2碳原子反应性较低,不足以促进氢的吸附,因此打造氢吸附的活性位是首要任务。Gohari-Bajestani等[16]也指出,可通过金属或金属氧化物等功能纳米粒子在石墨烯表面上的修饰来增强石墨烯衬底的气体吸附性能。
石墨烯是由碳原子以sp2杂化轨道组成六角型的二维薄膜,具有高比表面积、氢溢效应和结构多样性等特点,易于修饰和改性。在现有研究工作中,多作为金属、金属氧化物及高分子材料的载体进行氢气吸附[17-19],从宏观层面对石墨烯吸附气体进行了研究。但其应用仍存在一定的局限性,缺乏对石墨烯本身吸附氢气过程微观机理的考虑,无法对吸附现象进行微观过程的描述和分析:如氢气分子在石墨烯结构中的吸附位置与构型、利用化学氧化法对石墨烯进行表面改性过程中引入的一些含氧官能团,如烃基、羧基、和环氧基等对吸附反应过程的影响等,造成石墨烯材料选择因素的单一性,是目前石墨烯材料发展过程中亟需面对的问题。
通过计算机模拟计算,可以在原子尺度上一定程度地说明气体在吸附材料表面的微观吸附机理,是研究气体在石墨烯表面的吸附性质及表面微观吸附机理的有效手段。笔者构建出原始石墨烯及具有烃基、环氧基、边缘羧基、边缘羰基修饰的石墨烯分子模型,利用Material Studio 2019软件,研究了石墨烯对氢分子的吸附行为和性能及其表面含氧官能团对氢气吸附过程中吸附作用能、吸附构型和吸附等温线、吸附热的微观影响,从微观层面观察了氢分子在修饰不同含氧官能团石墨烯中的吸附过程,并直观地观察到氢分子的吸附状态和吸附位。研究结果可为选择和开发吸附性能优异的以石墨烯为基底的吸氢材料提供理论基础和结构设计依据。
1 模拟计算
1.1 模型的构建
首先通过Amorphous Cell模块构建原始石墨烯表面模型,将石墨烯结构单元填充到(2.95 nm×2.55 nm×3.00 nm (x×y×z))三维周期性边界晶胞盒子中得到石墨烯表面模型,其碳原子数量为222个,如图1(a)所示;同时构建氢气分子模型,如图1(b)所示。为了研究吸附过程中含氧官能团的影响,根据Lerf-klinowski模型[20-21],即羟基和环氧基随机分布在氧化石墨(Graphite oxide,GO)表面,羰基和羧基分布在GO边缘,构建了带有羟基、环氧基、边缘羰基、边缘羧基4种含氧官能团的石墨烯分子模型,分别记为C—OH、C—O—C、C=O和C—COOH,如图1(c)~(f)所示。4种单一含氧官能团占比均为15%,数量为45个,随机分布于GO上下表面。忽略因添加含氧官能团造成应力分布不均而导致的石墨烯片层弯曲与褶皱,设置刚性碳骨架结构,利用Forcite模块对模型结构进行优化和退火处理,以保证构建的模型具有最低能量和最优构型[22],将几何优化结果与相关文献进行对比:其中石墨烯的C—C键长为0.1420 nm,H—H键长为0.0741 nm,与文献报道的C—C的键长0.1421 nm[23],H—H键长0.0730 nm[24]相近,说明了所构建模型的合理性。
1.2 分子动力学模拟
分子动力学模拟在Materials Studio 2019软件上进行,计算过程采用基于Ewald加和与Atom方法计算的COMPASS力场[25],通过Forcite模块中的Geometry optimization和Anneal任务进行结构优化和退火处理,精度均设为Fine,退火温度范围为300~500 K,时间步长为1.0 fs,总模拟时间为0.5 ns;范德华相互作用采用Ewald求和法,截断半径为1.25 nm,精度为4.186×10-4kJ/mol,得到优化后的石墨烯表面模型。在不同含氧官能团石墨烯表面模型中加入单个H2分子,构成Graphene+H2、C—OH+H2、C—O—C+H2、C=O+H2、C—COOH+H2体系,重复上述结构优化方法对5个体系分别进行结构优化。动力学性质计算在Forcite模块中的Dynamics任务下进行,采用NVT正则系综,初始速度服从玻尔兹曼随机分布,温度分别为77、90、112、298 K,总模拟时长0.5 ns,时间步长为1.0 fs,温度控制采用Nose方法,范德华相互作用采用Ewald求和方法,截断半径为1.25 nm,精度为4.186×10-4kJ/mol。
1.3 巨正则蒙特卡洛模拟
采用巨正则蒙特卡洛模拟方法计算了氢气吸附等温线和吸附热,在Materials Studio 2019软件中的Sorption模块中进行。首先对石墨烯表面模型进行结构优化和退火处理,选取COMPASS力场,计算精度设置为Fine,范德华相互作用采用Ewald&Group求和法,截断半径为1.25 nm,精度为4.186×10-4kJ/mol,温度分别设置为77、90、112、298 K,压力选取范围为1×10-6~1 kPa,将氢气作为吸附质计算其在石墨烯表面的吸附等温线。分子之间的相互作用采用范德华力和库仑力综合作用进行描述的Lennard-Jones (LJ)12∶6势能模型[26]:
(1)
其中,σij和εij可根据L-B混合规则得:
(2)
式中:rij为2个相互作用原子i、j间距离,nm;εij为2个相互作用原子i、j间势能,其最小值称为相互作用的势能阱深,kJ/mol;σij表示2个相互作用原子i、j之间作用为零时的距离,常称为特征直径或碰撞直径,nm;qi、qj分别为原子i、j所带的电荷,C;ε0为介电常数,F/m;σi、σj、εi、εj代表原子i、j的非键参数;i、j为任意2个不同的原子。
2 模拟结果与分析
2.1 吸附构型和吸附作用能
带有不同含氧官能团的石墨烯模型在温度77~298 K下氢气吸附构型如图2所示。由图2可知,氢气分子均近似平行地吸附在石墨烯片层的较边缘位置,氢气分子和石墨烯片层的结构几乎没有发生变化,说明二者之间仅存在范德华力,即只进行了物理吸附[27-28]。
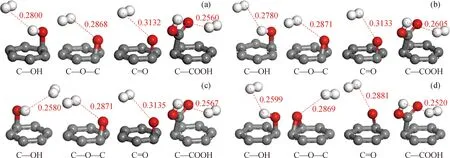
The red number represents the minimum distance between H2 and each oxygen-containing functional group structural unit at the most stable sorption configuration,the unit is nm.
为了更清楚地展示分子间距的变化,列出了氢气分子与带有不同含氧官能团石墨烯最稳定吸附时,氢气与各含氧官能团结构单元间的最小距离如表1所示。由表1可知,在不同温度下,氢气分子与不同含氧官能团之间的最小距离由大到小排序为C=O>C—O—C>C—OH>C—COOH。在不同温度下,氢气分子与含氧官能团间的吸附构型和最小距离变化不大。氢气与各含氧官能团结构单元的最小距离与氢分子的动力学直径0.296 nm相近[29],说明氢气可以吸附在表面有含氧官能团的石墨烯上,且氢气分子吸附于其表面的最有利位置为距固体表面约1个分子动力学直径的位置,即吸附位中的最小势能处。若以六元环中带有含氧官能团的碳原子为1号位,其余碳原子以顺时针方向分别命名为2号位~6号位(其中环氧基连接的2个碳原子以1号位起始,6号位结束),则可以看出,对于添加烃基和羰基的石墨烯,氢气分子的吸附靠近5号碳原子;对于添加环氧基和羧基的石墨烯,氢气分子的吸附靠近3号碳原子。可见不同的含氧官能团会对石墨烯的电荷分布和化学势能等产生影响,继而造成氢气分子吸附的不同趋向。

表1 H2与带有含氧官能团石墨烯结构单元间的最小吸附距离
吸附作用能可以用来研究吸附剂基底与氢气分子之间的相互作用强度,相互作用能通过式(3)进行计算:
Ead=EH2/sheet-Esheet-EH2
(3)
式中:Ead为系统的吸附能,kJ/mol;Esheet、EH2和EH2/sheet分别为吸附剂单层的能量、H2分子的能量和吸附H2后吸附系统的总能量,kJ/mol。
根据定义计算得到不同温度下氢气分子在带有含氧官能团石墨烯表面最稳定吸附位点的吸附能如表2所示。由表2可知:氢气与石墨烯及表面有含氧官能团石墨烯间的相互作用能均为负值,说明吸附过程是自发进行的放热过程,吸附后体系更加稳定,降低温度有利于气体的吸附;相互作用能的绝对值越大,吸附作用越明显。氢气与带有含氧官能团石墨烯之间的吸附能绝对值间相差不大,其中C—COOH与氢气的吸附能绝对值最高,同时吸附稳定性可用吸附能的高低来表示,吸附能越低,吸附越稳定。计算结果表明,C—COOH对氢气显示出稍强的吸附能力,但对氢气的吸附稳定性相对较差。

表2 H2在带有不同含氧官能团石墨烯上的吸附能
2.2 吸附等温线和吸附热
通过巨正则蒙特卡洛模拟,计算了温度77~298 K下在压力1×10-6~1 kPa范围内石墨烯及带有不同含氧官能团石墨烯对氢气的吸附等温线,如图3所示。由图3可以看出,在77、90、112、298 K下的吸附等温线均为直线型吸附曲线,随着压力的升高,氢气吸附量一直处于上升状态。氢气为非极性分子,在吸附时不易形成氢键,因此氢气与石墨烯及带有不同含氧官能团石墨烯间的相互作用力比较弱。在压力1×10-6~1 kPa范围内,温度为77、90、112、298 K下,原始石墨烯的最大氢气吸附量分别为5.787、4.630、4.051、1.501 mmol/g,添加烃基石墨烯(C—OH)的最大氢气吸附量分别为4.738、4.032、3.080、1.173 mmol/g,添加环氧基石墨烯(C—O—C)的最大氢气吸附量分别为4.551、3.833、3.354、1.194 mmol/g,添加羰基石墨烯(C=O)的最大氢气吸附量分别为5.029、4.071、3.352、1.289 mmol/g,添加羧基石墨烯(C—COOH)的最大氢气吸附量分别为3.649、3.284、2.372、0.839 mmol/g。综上,不同温度下,原始石墨烯对氢气的吸附量均为最大,说明含氧官能团的添加不利于石墨烯对氢气的吸附;添加羧基(C—COOH)的石墨烯模型对氢气吸附量及吸附速率均最低,说明了羧基的添加更不利于氢气的吸附。随着温度的降低,吸附量增大,这是因为温度的升高使氢气分子具有较大的动能[30],需要更大的吸附势能使氢气吸附作用大于其脱附作用,保证氢气在石墨烯上的吸附,因此,低温下石墨烯对氢气具有更好的吸附效果。这一结论与2.1节中对氢气分子与吸附剂间吸附作用能的计算分析结果相同。
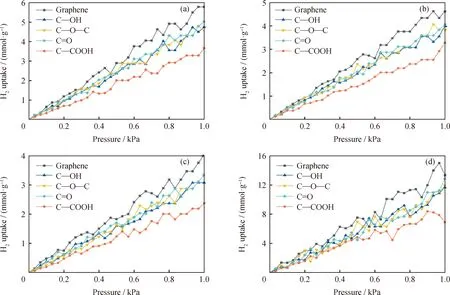
图3 不同温度下H2在石墨烯表面模型上的吸附等温线
吸附热是指吸附过程产生的热效应,对吸附有着重要的影响。吸附热的大小从某种程度上说明吸附质解吸的难易程度,吸附热越小,越容易解吸。吸附热通过式(4)进行计算。
(4)
式中:Qsf为吸附热,J/mol;p为吸附压力,Pa;R为气体常数,J/(mol·K);T为吸附温度,K。
在1×10-6~1 kPa压力范围内、不同温度下石墨烯及带有不同含氧官能团石墨烯对氢气的吸附热曲线如图4所示,计算不同温度下石墨烯及带有不同含氧官能团石墨烯对氢气吸附的平均吸附热,结果列于表3。由图4可知,原始石墨烯及带有不同含氧官能团石墨烯的吸附热数据在同一温度下随压力变化的相对平均偏差最大值为4.813%,波动幅度较小,近似恒定不变的吸附热对应于图3中的直线型吸附等温线,表明压力的变化对于氢气在石墨烯中的吸附热影响较小。若吸附剂表面吸附能量均一,则吸附质分子之间并无作用力,吸附热也与吸附剂的表面覆盖率无关。而较低的吸附热则表明,石墨烯上并没有对氢气分子产生特殊吸附的位置,因此在低压下提高氢气吸附量的主要方法是打造石墨烯表面活性位,产生有效的吸附位点,改善其对氢气分子的相互作用能,这一点与Eftekhari等[15]的研究相一致。
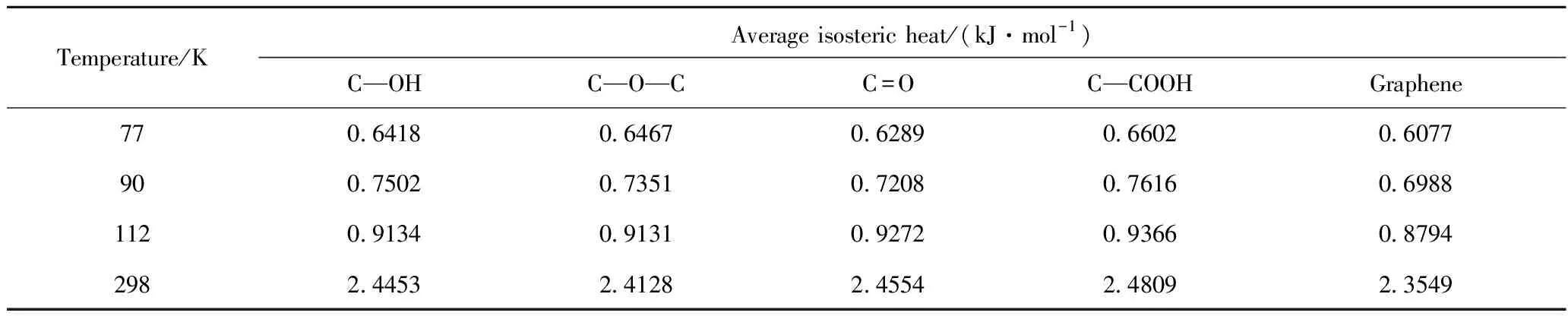
表3 不同温度下H2在石墨烯表面模型上的平均吸附热
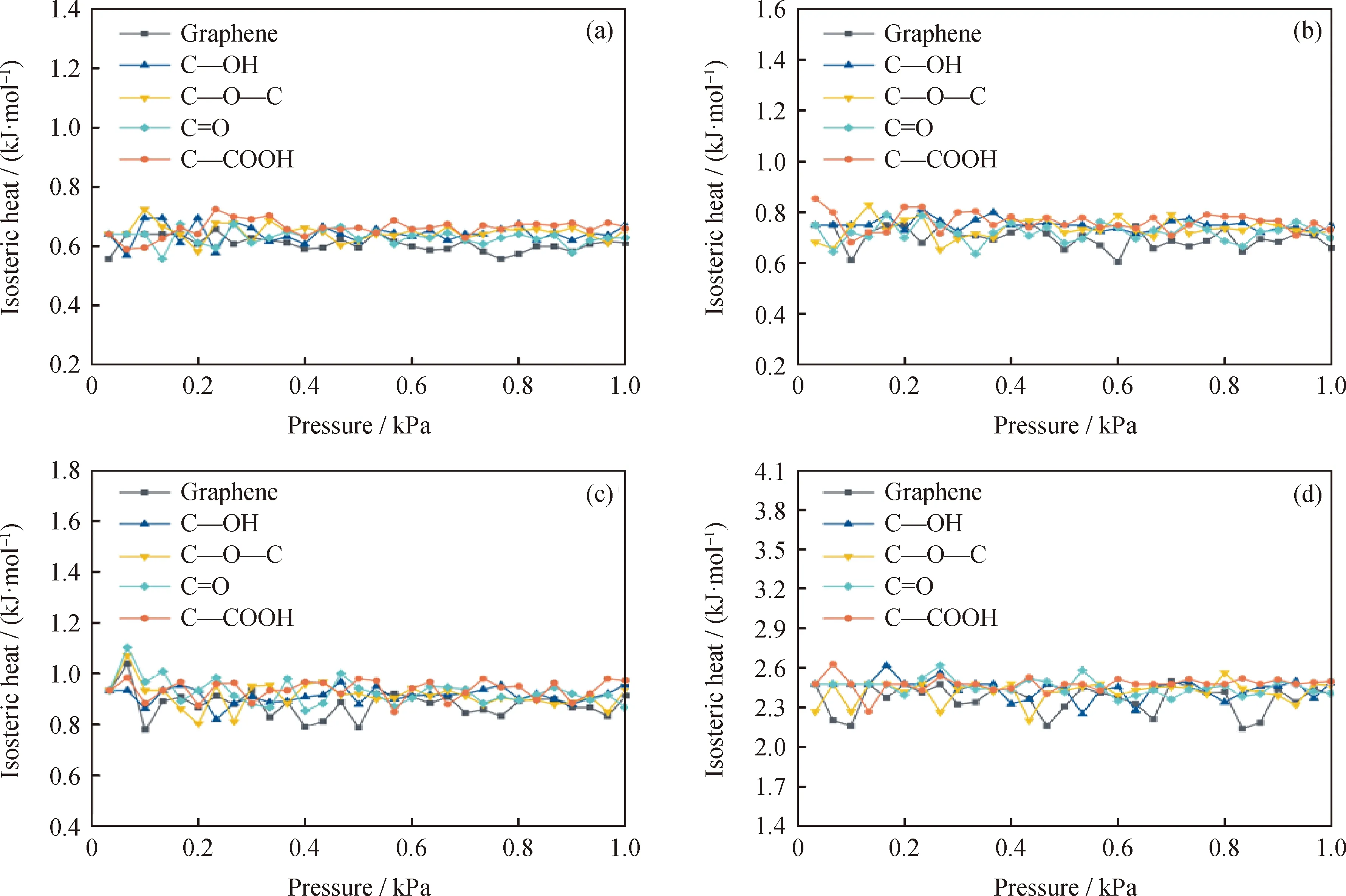
图4 不同温度下H2在石墨烯表面模型上的吸附热曲线
由表3可以看出,在压力1×10-6~1 kPa范围内,不同温度下带有不同含氧官能团石墨烯对氢气的平均吸附热中,C—COOH的平均吸附热均为最大;在温度77和90 K下,C=O的平均吸附热最小,在温度112和298 K下,C—O—C的平均吸附热最小。这与氢气在带有不同含氧官能团氧化石墨烯模型中的吸附等温线变化并不一致,说明在石墨烯的氢气吸附过程虽为物理吸附,但吸附过程仍然非常复杂,吸附量与吸附热的大小与吸附剂和吸附质的性质等其他因素都有关。
3 结 论
构建了石墨烯及带有不同含氧官能团的石墨烯模型,采用分子动力学及蒙特卡洛模拟方法,分析氢气分子在其表面的吸附行为。对石墨烯吸附氢气过程中的吸附作用能、吸附构型和吸附等温线、吸附热进行了分析,研究了石墨烯表面不同含氧官能团对其吸氢性能的影响。主要结论如下:
(1)由石墨烯及表面带有含氧官能团石墨烯与氢气的吸附构型可知,二者之间仅存在范德华力,即只进行了物理吸附;在不同温度下,氢气分子与含氧官能团间的吸附构型和最小距离变化不大;氢气与各含氧官能团结构单元的最小距离与氢分子的动力学直径0.296 nm相近;不同的含氧官能团会造成氢分子吸附的不同趋向。
(2)氢气与石墨烯及表面带有含氧官能团石墨烯间的相互作用能均为负值,吸附过程是自发进行的放热过程,降低温度有利于气体的吸附;氢气与带有含氧官能团石墨烯之间的吸附能绝对值间相差不大,其中C—COOH与氢气的吸附能绝对值最高。
(3)在温度77~298 K、压力1×10-6~1 kPa范围内,氢气与石墨烯及表面带有含氧官能团石墨烯间的吸附等温线均为直线型吸附曲线,随着压力的升高,氢气吸附量一直处于上升状态;4种温度下,原始石墨烯对氢气的吸附量和吸附速率均为最大,添加羧基(C—COOH)的石墨烯模型对氢气的吸附量及吸附速率均最低,含氧官能团的添加不利于石墨烯对氢气的吸附。
(4)近似恒定不变的吸附热对应于直线型吸附等温线,表明温度与压力的变化对于氢气在石墨烯中的吸附热影响较小;较低的吸附热表明,石墨烯上并没有对氢气分子产生特殊吸附的位置,因此在低压下提高氢气吸附量的主要方法是打造石墨烯表面活性位,产生有效的吸附位点。
(5)通过分子模拟分析了石墨烯表面不同含氧官能团对氢气吸附特性的影响规律,结果表明,在氢气吸附基底材料的选择中,氧化石墨烯所带有的含氧官能团不利于氢气吸附,材料制备中应通过还原手段将其除去,并通过在石墨烯上打造氢气吸附的活性位以获得更好的吸氢效果。研究工作能够指导对石墨烯针对性表面改性及对其功能化修饰,使之与应用基体相匹配,以发挥更好的吸附性能。
猜你喜欢
中学化学(2022年5期)2022-06-17
中学生数理化·高一版(2022年4期)2022-05-09
中国特种设备安全(2021年4期)2021-10-13
中学生数理化(高中版.高考数学)(2020年2期)2020-04-21
中学生数理化·高二版(2016年3期)2016-12-26
中学生数理化·高二版(2016年3期)2016-12-26
材料科学与工程学报(2016年5期)2016-02-27
高中生学习·高二版(2015年3期)2015-05-21
中学政史地·教学指导版(2014年10期)2015-02-02
发明与创新(2013年13期)2013-03-11
