
多孔碳材料室温下催化氧化H2S的研究进展
2023-11-01 08:19:26刘晓丽李建军
天然气化工—C1化学与化工 2023年5期
刘晓丽,李建军,李 新
(1.四川大学 建筑与环境学院,四川 成都 610065;2.四川大学 碳中和未来技术学院,四川 成都 610065;3.成都达奇科技股份有限公司,四川 成都 610065)
硫化氢(H2S)是一种无色、有剧毒、腐蚀性、易燃性和臭鸡蛋气味的酸性气体,广泛存在于石油裂解气、天然气、沼气、高炉煤气、焦炉煤气和转炉煤气等工业气体中。此外,部分含硫有机物存放时间过长,也会产生H2S[1-4]。在工业领域,即使是很少量的H2S,也会造成生产设备和输送管道的腐蚀,并且会导致催化剂的失活或中毒[5]。H2S 对人类健康也存在威胁,低浓度(无特别说明本文均指体积分数,小于30 × 10-6)的H2S 即可损害人体的呼吸、神经系统,而高浓度(大于700 × 10-6)的H2S会致人死亡[6]。此外,H2S在空气中会被氧化成硫氧化物,诱发酸雨或雾霾,破坏生态环境。因此,开发从工业气体中高效、经济地脱除H2S的方法至关重要。
目前,H2S 的脱除方法主要有吸收法[7]、吸附法[8]、生物法[9]和催化氧化法[10]。吸收法一般采用乙醇胺、低温甲醇或氢氧化钠溶液等作为吸收剂[11],具有处理气量大、适用高浓度H2S的优点,但存在脱硫精度低的问题,常用于粗脱硫[12]。吸附法具有脱硫精度高、操作简单等优点,适用于处理气量小、低浓度H2S 的处理,常用于精脱硫[13]。生物法操作条件温和、成本低,但技术尚不成熟,不能处理高浓度H2S[14]。克劳斯法是一种处理高浓度H2S、回收硫磺的经典催化氧化法,具有处理气量大、适用高浓度H2S和装置简单等优点,但由于受热力学平衡限制,两级反应器装置的典型硫回收效率仅为90%~95%,导致尾气中含硫过高、硫磺回收装置堵塞等。此外,该方法的操作温度较高导致能耗高[15-16]。而室温下催化氧化法具有操作条件温和、脱除效率高、适用低浓度H2S、可获得单质硫,以及无热力学平衡限制等优点,被认为是非常有应用潜质的H2S 脱除方法[17]。开发具有高选择性、高硫容量、高脱硫活性和可循环再生的催化剂是室温下催化氧化H2S的核心。目前,研究较多的催化剂主要有金属氧化物[18-19]、分子筛[20-21]和多孔碳材料[22-23]等。其中,多孔碳材料因其比表面积高、孔结构发达、易于表面改性和化学性质稳定等优点而吸引了越来越多研究者的关注。一般来说,多孔碳材料的孔隙结构、比表面积和表面化学性质等均会影响其室温下催化氧化H2S的性能(脱硫性能)。PAN等[24]发现室温下具有孔径为0.40 nm左右超微孔的多孔碳材料可以将O2活化为超氧化物自由基(·O-2)。吉布斯自由能计算结果显示,在孔径大于0.43 nm 的孔中O2吸附是热力学有利的;当孔径小于0.39 nm时,效果是相反的;在孔径为0.40~0.42 nm 的孔中吉布斯自由能几乎为零,表明O2分子在超微孔中的吸附不受限制,并且很容易从超微孔中解吸,保证有充足的·O-2参与H2S的催化氧化。用碱性物质浸渍多孔碳材料可提高其对应所得催化剂的碱性,进而促进H2S 的吸收和解离[25]。然而,碱性物质的快速消耗会导致催化剂的脱硫性能急剧降低,而因失活催化剂产生的固体废物可能造成二次污染。对于多孔碳材料来说,氮原子掺杂是较好的改性方法,因为氮原子上分离的电子对可以替代碱性物质作为碱性位点[26]。在脱硫过程中,含硫产物容易沉积在催化剂的微孔中,逐渐堵塞孔道并消耗催化活性位点,导致催化剂失活。因此,设计并开发价格低廉、稳定性高和使用寿命长的催化剂,是近年来研究的重点,也是多孔碳材料用于室温下催化氧化H2S 的最大挑战。
本文结合国内外近年来的研究成果,首先对多孔碳材料室温下催化氧化H2S 的反应机理进行分析,然后在此基础上对室温下催化氧化H2S 的多孔碳材料的研究进展进行综述,最后对其未来的发展方向进行展望,以期为室温下催化氧化H2S 的多孔碳材料的研发和工业应用提供理论支撑。
1 多孔碳材料室温下催化氧化H2S机理
多孔碳材料室温下催化氧化H2S是一个非常复杂的过程,通常包括物理吸附、化学吸附和催化氧化这3步。多孔碳材料室温下催化氧化H2S的机理见图1[17]。具体来说,气相中的水蒸气吸附在多孔碳材料上,在其表面形成一层水膜;气相中的H2S和O2扩散后被吸附进入多孔碳材料的孔隙内,随后溶解在水膜中;H2S解离成HS-,同时吸附的O2被活化为O*,HS-与活性氧物种在多孔碳材料-水膜界面处发生反应生成单质硫。少量单质硫会被进一步氧化成SO2,在有O2和水的情况下形成硫酸[27]。O2被活化和H2S解离被认为是室温下催化氧化H2S的关键步骤。
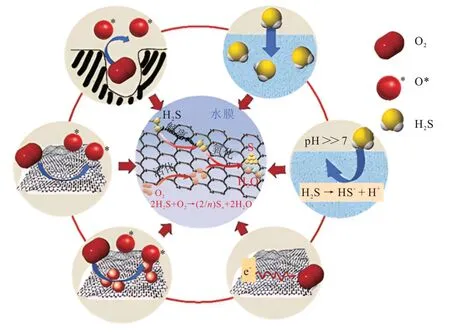
图1 多孔碳材料催化氧化H2S的机理示意[17]Fig.1 Mechanism schematic diagram of catalytic oxidation of H2S by porous carbon materials[17]
2 多孔碳材料室温下催化氧化H2S 研究进展
近年来,研究人员针对活性炭、介孔炭、碳纳米管、石墨烯、碳纤维和MOFs等多孔碳材料在室温下催化氧化H2S 的性能开展了大量研究,取得丰富的成果。以下分别就活性炭、介孔炭、碳纳米管、石墨烯和碳纤维这5种多孔碳材料展开详细论述。
2.1 活性炭
活性炭(AC)是最常见的多孔碳材料,因其具有价廉易得、比表面积较大、孔隙结构较发达和机械稳定性较好等优点而被广泛应用于工业脱除H2S过程。然而,由于活性炭的孔隙一般为微孔,且表面具有疏水性不利于H2S的吸附,导致脱硫性能较差。通过杂原子(N、P 或S)掺杂、碱性溶液浸渍或金属氧化物负载等方法对活性炭进行改性,可以提高其催化氧化H2S 的性能。近年来,不同活性炭催化氧化H2S的相关数据见表1。
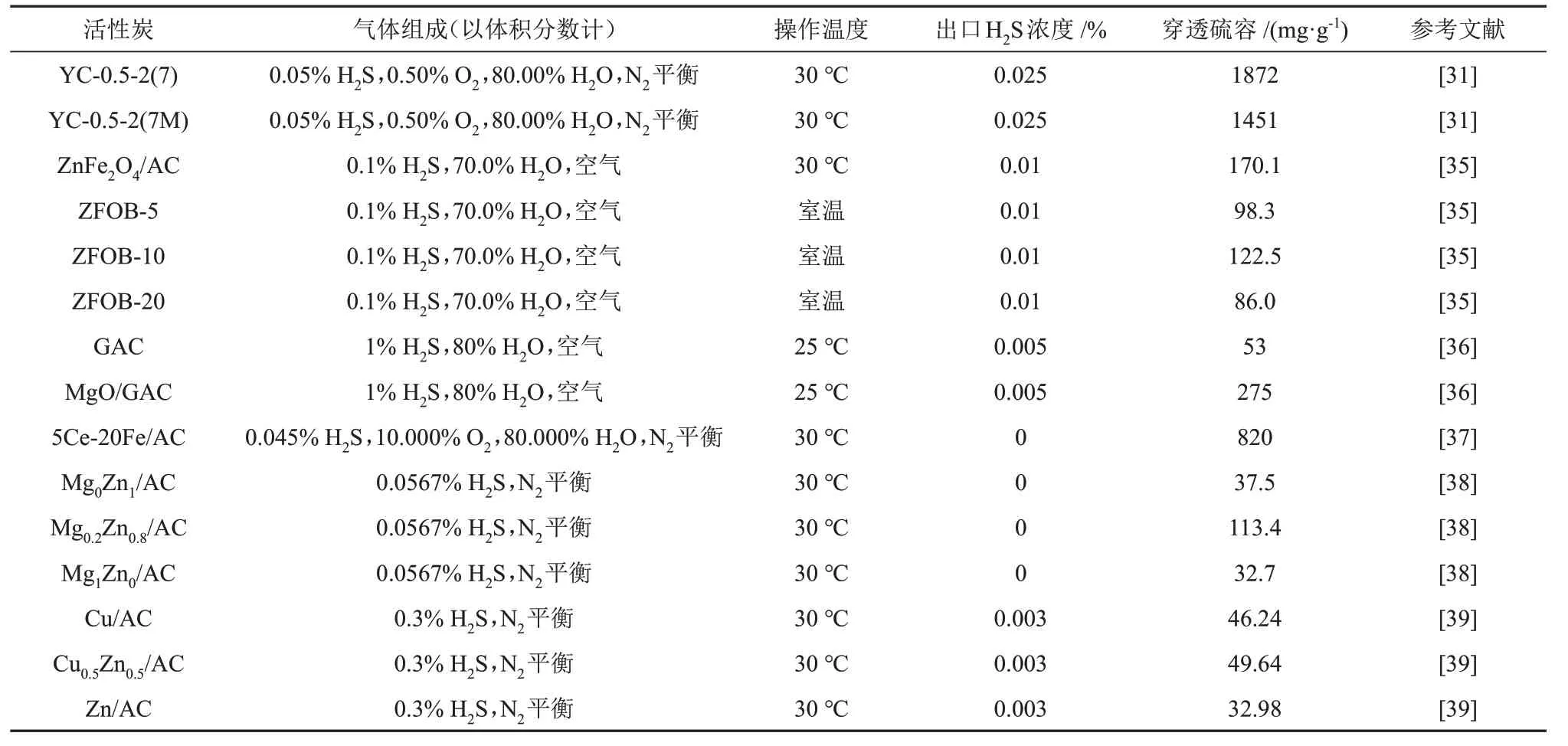
表1 不同活性炭催化氧化H2S的相关数据Table 1 Relevant data of catalytic oxidation of H2S by different activated carbons
LⅠ等[28]研究了活性炭的比表面积、孔隙结构和表面酸碱性对其催化氧化H2S 性能的影响,发现酸性条件有利于H2S 转化为硫酸盐,碱性条件有利于单质硫的形成。在活性炭表面引入杂原子,可以定向调控活性炭的表面电子结构和酸碱性[29]。氮原子是目前常用的活性炭表面掺杂原子之一,主要通过含氮前驱体(三聚氰胺、脲等)引入活性炭中,以增加碱性官能团的含量。含氮官能团尤其是吡啶氮、吡咯氮和季氮,可以促进O2的化学吸附、活化以及HS-的解离,进而提高活性炭的脱硫性能[30]。WU 等[31]以超分子三聚氰胺氰尿酸作为氮源,采用一步热解法制备了氮含量超高(17.2%,原子百分数)且具有分级孔结构的氮掺杂生物质衍生活性炭(NBAC),并用于室温下催化氧化H2S(图2)。作者通过掺杂氮原子引入了大量碱性含氮官能团和缺陷,为H2S 的催化氧化提供了活性位点,有助于H2S 和O2的解离,同时微孔结构促进了反应物和单质硫的传递,穿透硫容达1827 mg/g。利用NaOH、KOH 和Na2CO3溶液等碱性溶液浸渍活性炭也是常用的表面改性方法[32-33],经过碱性溶液的浸渍改性,活性炭的穿透硫容可较普通活性炭提高3~29倍[34]。
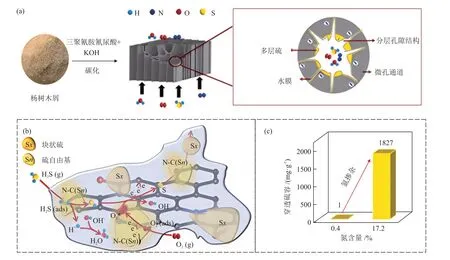
图2 NBAC的制备过程示意(a)、H2S的去除机理(b)和NBAC在室温下催化氧化H2S的性能(c)[31]Fig.2 Schematic diagram of preparation process of NBAC (a), mechanism of H2S removal (b) and catalytic oxidation performance of H2S by NBAC at room temperature (c)[31]
另外,对活性炭进行金属(Zn、Cu、Fe和Mg等)改性可以产生更多的活性中心,大幅提高其催化氧化H2S的性能。YANG等[35]采用浸渍法制备了一系列不同ZnFe2O4负载量(质量分数,下同)的ZnFe2O4/活性炭,并分别用于室温下催化氧化H2S,部分实验结果见图3。作者发现,ZnFe2O4/活性炭的穿透硫容随ZnFe2O4负载量的增加呈现先增大后减小的趋势,当ZnFe2O4的负载量为10%时,在室温、含水量(体积分数,下同)为70%的条件下脱除H2S 时,ZnFe2O4/活性炭的穿透硫容达到最大值(122.5 mg/g)。随着ZnFe2O4负载量的增加,活性炭上活性位点增加,但当ZnFe2O4的负载量过大时,会堵塞活性炭的孔道、比表面积降低,进而影响脱硫性能。脱硫过程中形成的FeOOH 中间体对H2S 的催化氧化起到了较明显的促进作用,经过3次再生后,仍保持较高的脱硫性能。SⅠRⅠWARDANE等[36]采用浸渍法将纳米MgO 引入到活性炭的微孔中制得MgO/活性炭。当纳米MgO的负载量为1.2%时,MgO/活性炭的穿透硫容达到275 mg/g,较原始活性炭(53 mg/g)提高了5.18倍。在脱硫过程中,H2S在MgO/活性炭上同时发生物理吸附和化学吸附,物理吸附主要发生在活性炭的介孔结构上,化学吸附主要发生在MgO位点上。纳米MgO不仅是催化氧化H2S的活性中心,还为H2S化学吸附提供了活性位点。
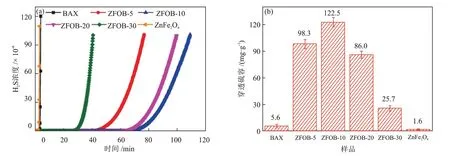
图3 ZnFe2O4/活性炭的穿透曲线(a)和对应的穿透硫容(b)[35]Fig.3 Breakthrough curves (a) and corresponding breakthrough sulfur capacities (b) of ZnFe2O4/activated carbon[35]
除了单金属掺杂,越来越多的学者尝试将双金属掺杂到活性炭中,以期发挥双金属协同效应的优势。LⅠU 等[37]采用浸渍法制备了Ce-Fe/活性炭,并用于室温下催化氧化H2S,发现掺杂Ce显著提高了穿透硫容和延长了穿透时间,分别达到820 mg/g和71 h。CeO2储存氧和释放氧的能力促进了脱硫过程中Fe2+氧化为Fe3+,降低了催化氧化H2S时对O2的需求,从而增强了脱硫性能。YANG 等[38]采用浸渍法制备了一系列MgO-ZnO/AC,用于室温下催化氧化H2S,并研究了保持金属氧化物总的负载量(20%)不变,调变MgO 与ZnO 的比例对MgO-ZnO 协同作用的影响。结果表明,原始活性炭几乎不具备H2S 脱除性能,而在负载ZnO 后,穿透时间为124 min,是原始活性炭的12 倍。继续引入MgO 后,穿透时间逐渐增加,当n(Mg)/n(Mg + Zn) = 0.2 时,穿透时间达到最大值(380 min)。随着n(Mg)/n(Mg + Zn)进一步增加,脱硫性能反而下降。其中Mg0Zn1/AC和Mg0.2Zn0.8/AC 的穿透硫容分别为38.5 mg/g 和113.4 mg/g。引入MgO 增加了活性炭的碱度,促进了HS-在ZnO 表面的化学吸附与单质硫的形成。FALCO 等[39]研究了同时使用ZnO 和CuO 改性对活性炭催化氧化H2S性能的影响。当Cu和Zn的物质的量相同时,Cu0.5Zn0.5/AC的脱硫性能优于单独负载Zn 或Cu 的催化剂,表明两种金属氧化物之间存在明显的协同效应,Cu 可以有效防止富Zn 催化剂催化氧化H2S时微孔的堵塞。
2.2 介孔炭
在催化氧化H2S 的过程中,含硫产物易在活性炭表面上沉积,导致微孔堵塞,进而使催化剂失活。由于介孔炭具有较大的孔隙,可为含硫产物的沉积提供充足的空间,因此可能具有较好的H2S 脱除性能。YU 等[40]以微孔Zn 基沸石咪唑骨架(ZⅠF-8)多面体为碳前体,通过熔盐法制得二维氮掺杂介孔炭纳米片。WANG 等[41]以聚乙烯亚胺为氮源,使用浸渍法制得氮掺杂介孔炭纳米片。CHEN等[42]分别以柏树木屑为碳前体、氮化碳为氮源、K2CO3为活化剂制备了氮掺杂介孔炭,并用于室温下催化氧化H2S。近年来不同介孔炭催化氧化H2S的相关数据见表2。
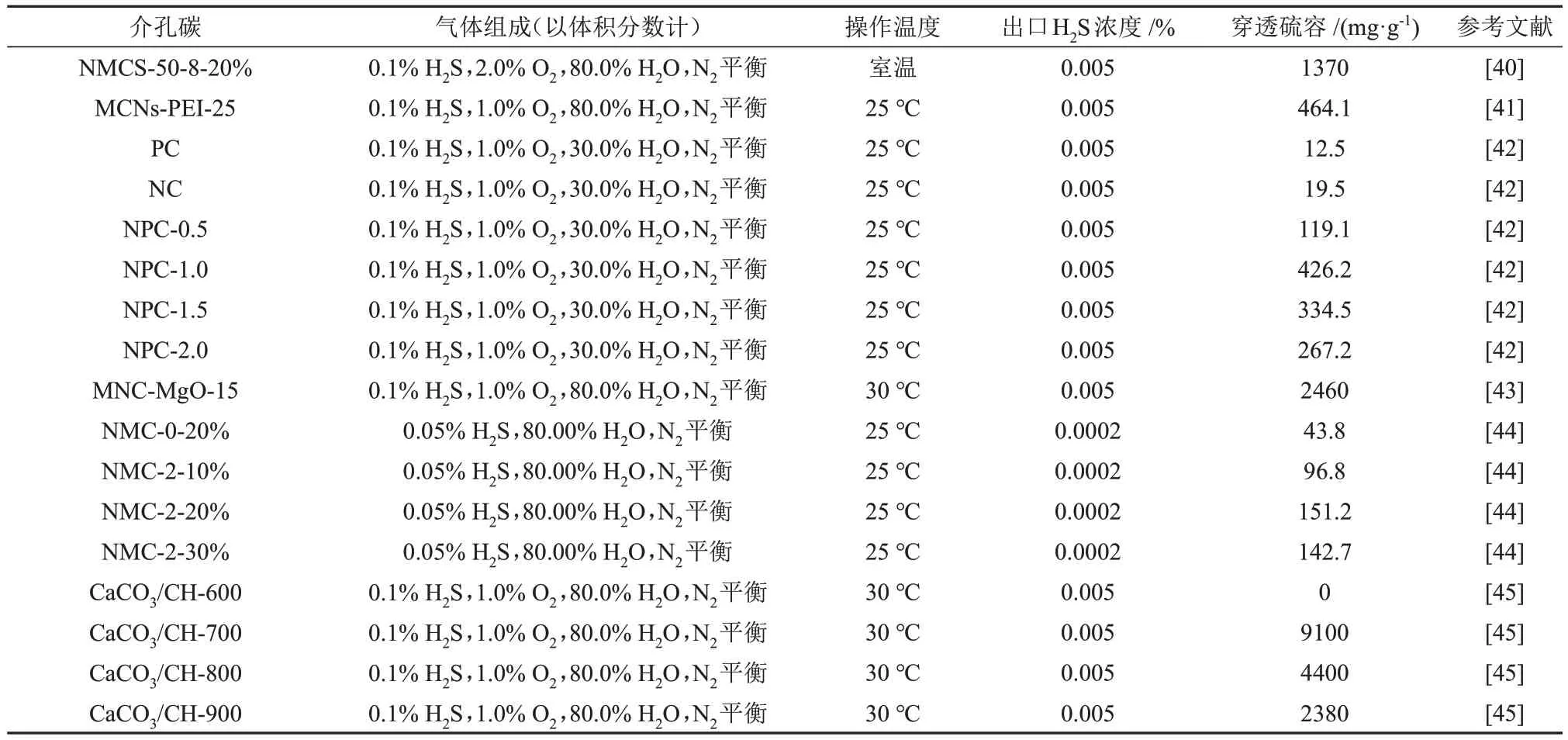
表2 不同介孔炭催化氧化H2S的相关数据Table 2 Relevant data of catalytic oxidation of H2S by different mesoporous carbons
ZHANG 等[43]采用悬浮辅助纳米塑形法制得毫米级介孔碳球(MCS),并进一步用碱性溶液(溶质分别为MgO、Na2CO3、NaOH、KOH 和K2CO3)对其进行浸渍改性,并用于室温下催化氧化H2S,不同碱性溶液浸渍和不同MgO 负载量的MCS 的穿透曲线见图4。
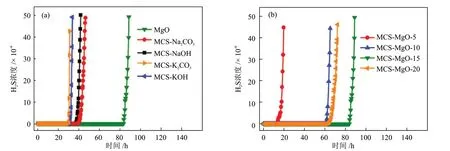
图4 不同碱性溶液浸渍(a)和不同MgO负载量(b)的MCS的穿透曲线[43]Fig.4 Breakthrough curves of MCS impregnated with different alkaline solutions (a) and with different MgO loadings (b)[43]
作者发现,不同碱性溶液浸渍对MCS的脱硫性能的改善效果由高到低依次为:MgO溶液、Na2CO3溶液、NaOH溶液、KOH溶液和K2CO3溶液。用MgO溶液浸渍后的MCS的穿透硫容随MgO负载量的增加呈现先增大后减小的趋势,当MgO负载量为15%时,在反应温度为30 ℃、含水量为70%的条件下,用MgO溶液浸渍后的MCS的穿透硫容达到最大值(2460 mg/g),远大于市售活性炭(400~600 mg/g)。由于MCS 交互连通的介孔有利于H2S 和产物的扩散,单质硫不会聚集或堵塞孔隙,MgO可溶于水膜并不断释放OH-,提高了脱硫性能。
CHEN 等[44]采用有机-无机共组装法制得具有大比表面积(1172 m2/g)的氮掺杂有序介孔碳(NMC),并进一步添加双氰胺和负载ZnO对其进行改性,用于室温下催化氧化H2S。NMC的制备过程和不同双氰胺添加量、不同ZnO 负载量的NMC 的穿透曲线分别见图5(a)和图5(b)。作者发现,ZnO负载量(20%)相同时,双氰胺的添加量为2 g 的NMC-2-20%的穿透硫容为151.2 mg/g,是双氰胺的添加量为0 g 的NMC-0-20%的3.47 倍,吡啶N 作为Lewis碱性位点,增加了碱性吸附位点的数量,促进了H2S的解离。当氮掺杂量相同时,NMC的穿透硫容随ZnO负载量的增加呈现先增大后减小的趋势,ZnO 负载量为20%时,穿透硫容达到最大值,ZnO增加了活性位点和吡啶N的数量,氮掺杂和ZnO之间的相互作用共同促进了H2S的催化氧化。
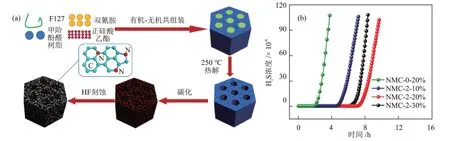
图5 NMC的制备过程示意(a)和穿透曲线(b)[44]Fig.5 Schematic illustration of the preparation (a) and breakthrough curves (b) of NMC[44]
PAN 等[45]以葡萄糖酸钙为碳前体,采用一步炭化法合成了具有二维纳米片结构的介孔CaO/碳异质结,二维CaO/碳异质结的制备过程、穿透曲线和对应的穿透硫容见图6。二维纳米片结构为催化氧化H2S 的反应提供了更多暴露的活性位点和更大的硫产物储存空间,具有良好表面化学性质和高度分散特性的CaO 纳米颗粒促进了活性氧自由基的形成和H2S 的解离,使制备得到的异质结表现出9100 mg/g 的超高穿透硫容。
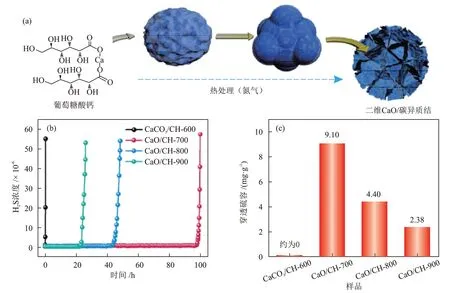
图6 二维CaO/碳异质结的制备过程示意(a)、穿透曲线(b)和对应的穿透硫容(c)[45]Fig.6 Schematic diagram of preparation process (a), breakthrough curves (b) and corresponding breakthrough sulfur capacities (c)of 2D CaO/carbon heterostructures[45]
2.3 碳纳米管
碳纳米管(CNTs)是一种以sp2杂化为主的碳材料,具有高纵横比和中空圆柱形态。根据石墨层的数量,分为单壁碳纳米管、双壁碳纳米管和多壁碳纳米管。由于CNTs 优异的物理化学性质,如中空和层状结构、π-共轭结构、侧壁曲率、高稳定性和导电性等,被认为是非常有潜力的室温下催化氧化H2S的材料[46]。
RASHⅠDⅠ等[47]比较了烯丙基酰胺接枝多壁碳纳米管(AGMWCNTs)和氧化多壁碳纳米管(OMWCNTs)的脱硫性能。由于胺基官能团的存在,AGMWCNTs表现出比OMWCNTs更高的硫容,胺基官能团为H2S 的解离提供了碱性环境,同时也是H2S化学吸附的活性位点。JⅠ等[48]采用共沉淀法将纳米水合氧化铁(HFO)锚定在多壁碳纳米管(MWCNTs)上,并测试了所得样品(FeC-x,x为MWCNTs 的质量分数)的低温催化氧化H2S 的性能,部分结果见图7。作者发现,MWCNTs几乎不具备脱硫性能,穿透硫容为0.76 mg/g,HFO 的穿透硫容为83.48 mg/g。添加MWCNTs 后,显著提高了HFO 的低温脱硫性能,随着MWCNTs 质量分数的增加,HFO 的脱硫性能呈现先提高后降低的趋势,当MWCNTs 质量分数为20%时,FeC-0.2 的穿透硫容达到最大值(144.2 mg/g)。因为高度分散的HFO纳米颗粒增加了活性位点,MWCNTs 与HFO 之间相互作用形成的Fe—O—C 键促进了MWCNTs 和HFO 之间的电子转移,进而提高了HFO 的脱硫性能,但过量的MWCNTs会降低单位质量脱硫剂中活性组分的含量。出毅能等[49]采用聚乙烯亚(PEⅠ)对MWCNTs进行功能化,发现MWCNTs-PEⅠ对H2S的脱除效果为0.16 mmol/g。红外光谱和热失重分析结果表明,PEⅠ成功键合到MWCNTs 上,拉曼光谱中碳纳米管的ID/IG值(D峰面积与G峰面积之比)从0.8398 变成1.2364,说明碳纳米管的缺陷程度明显增加,缺陷程度的增加是由碳纳米管表面引入PEⅠ导致的。
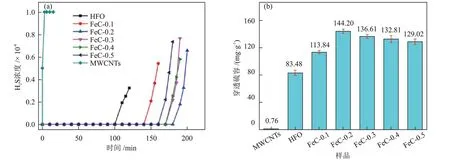
图7 MWCNTs、HFO和FeC-x的穿透曲线(a)和对应的穿透硫容(b)[48]Fig.7 Breakthrough curves (a) and corresponding breakthrough sulfur capacities (b) of MWCNTs, HFO and FeC-x[48]
2.4 石墨烯
石墨烯是碳的同素异形体之一,结构中包含由单原子层的sp2杂化碳原子组成的二维六边形晶格。石墨烯可以分为原始石墨烯、氧化石墨烯(GO)和还原氧化石墨烯(rGO)。石墨烯具有稳定的物理和化学性质、高电子迁移率和良好的热传递性能。然而原始石墨烯的性能并不能满足工业脱硫的要求,需通过碱性溶液浸渍或金属氧化物复载等方法对其进行改性,以提高其催化氧化H2S 的性能。近年来不同石墨烯催化氧化H2S的相关数据见表3。

表3 不同石墨烯催化氧化H2S的相关数据Table 3 Relevant data of catalytic oxidation of H2S by different graphenes
PAN 等[27]采用水热合成工艺制得石墨烯气凝胶,然后用Na2CO3溶液浸渍,通过自组装形成具有三维网状结构的石墨烯气凝胶AGA-y(y为Na2CO3溶液的质量分数),其制备过程见见图8(a)。未浸渍Na2CO3溶液时,AGA-0 为无互连的三维网状结构,其SEM 照片和TEM 照片分别见图8(b)和图8(e),AGA-0 的脱硫性能很差,穿透硫容可忽略不计,其穿透曲线和穿透硫容分别见图8(h)和图8(i)。当浸渍Na2CO3溶液后,样品的脱硫性能明显提高,并随着Na2CO3溶液质量分数的增加表现出先增加后降低的趋势,当Na2CO3溶液质量分数为30%时,AGA-30%的穿透硫容达3190 mg/g 或0.31 g/cm3(指体积为1.00 cm3的AGA-30%可容纳0.31 g H2S)。由于Na2CO3起到了交联剂的作用,表面含氧官能团通过氢键作用改善了碳酸盐在三维石墨烯气凝胶表面的分散度,三维石墨烯气凝胶提供大量的二维互连石墨烯片,极大地扩大了H2S的存储空间。当Na2CO3溶液的质量分数超过50%,样品的穿透硫容略有下降,其原因在于过量的Na2CO3会破坏石墨烯的三维网状结构,使石墨烯气凝胶形成棒状结构,AGA-75%的SEM照片和TEM照片分别见图8(d)和图8(g),石墨烯片含量的减少导致储存H2S的空间减少。
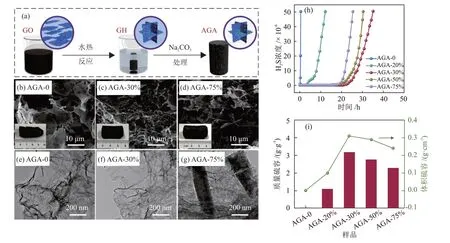
图8 AGA-y的制备过程示意(a)、SEM照片((b)~(d))、TEM照片((e)~(f))、穿透曲线(h)和对应的穿透硫容(i)[27]Fig.8 Schematic diagram of preparation process (a), SEM images ((b)~(d)), TEM images ((e)~(f)), breakthrough curves (h) and corresponding breakthrough sulfur capacities (i) of AGA-y[27]
XU 等[50]采用不同金属氧化物对rGO 进行改性,制备了一系列二维rGO 基复合材料,其制备过程、穿透曲线和对应的穿透硫容依次见图9(a)、图9(b)和图9(c)。
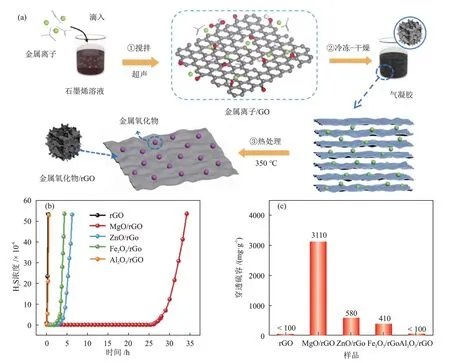
图9 金属氧化物/rGO的制备过程示意(a)、穿透曲线(b)和对应的穿透硫容(c)[50]Fig.9 Schematic diagram of preparation process (a), breakthrough curves (b) and corresponding breakthrough sulfur capacities (c)of metal oxide/rGO[50]
作者发现,二维rGO 基复合材料在室温下催化氧化H2S 的活性由高到低依次为:MgO/rGO、ZnO/rGO、Fe2O3/rGO、Al2O3/rGO≈rGO。未负载金属氧化物时,出口H2S浓度立即达到0.005%(图9(b)),rGO几乎不具有脱硫性能,主要是因为rGO的酸性含氧官能团会抑制H2S的吸附和解离。对于MgO/rGO,脱硫开始后25.0 h 内,出口H2S 浓度始终保持在0.00001%内,运行34.2 h 后才发生穿透,穿透硫容达3110 mg/g(图9(c))。超分散的纳米颗粒抑制了rGO 的堆积并保持其二维片状结构,为单质硫的存储提供了较大空间。此外,均匀的纳米颗粒为催化氧化反应提供了额外的碱性位点。作者通过密度泛函理论对MgO/rGO 催化氧化H2S 的反应过程进行了模拟,发现碱性MgO不仅可以促进H2S解离成HS-,而且具有捕获氧自由基的能力,使O2-可以停留更长时间,进一步促进HS-氧化成单质硫,MgO的消耗和活性位点的覆盖是复合材料失活的主要原因。在ZnO/rGO 和Fe2O3/rGO 上,HS-直接参与中和反应,分别生成金属硫化物ZnS 和Fe2S3,随着反应的进行,ZnO 和Fe2O3的连续消耗导致复合材料的脱硫性能不断降低,直至最终失活。Al2O3是一种两性金属氧化物,不能提供碱性位点,难以促进H2S的解离,因此,Al2O3/rGO 也几乎不具有脱硫性能。KHODADADⅠ[51]基于第一性原理计算后究发现,用金属Ni、Cu 或Zn 掺杂石墨烯可以增强其对H2S 的吸附。张文杰等[52]采用第一性原理方法对Pt 或Pt团簇修饰的石墨烯的几何结构、电子结构及其对H2S 的吸附、解离行为进行了研究。结果表明,H2S在Pt或Pt团簇修饰的石墨烯上均为弱物理吸附,但H2S 解离后的HS-和单质硫可以稳定吸附在材料表面,Pt掺杂石墨烯有利于吸附H2S,但不利于解离,Pt团簇掺杂的石墨烯能够轻松吸附并脱除H2S。
2.5 碳纤维
碳纤维因具有优异的性能,如高电导率、良好的机械性能和化学惰性,而成为具有吸引力的室温下催化氧化H2S的材料[53]。BAJAJ等[55]以聚丙烯腈/醋酸铜为前驱体,依次经过静电纺丝、炭化和活化处理,制备了负载Cu/CuxO纳米颗粒的碳纳米纤维。H2S 吸附动态穿透实验结果表明,负载Cu/CuxO 纳米颗粒的碳纳米纤维(938 min)的穿透时间比纯碳纳米纤维(62 min)延长了超过15倍,这主要是因为Cu/CuxO 纳米颗粒在碳纳米纤维表面上分散良好。SUN 等[55]分别以聚丙烯腈(PAN)和ZⅠF-8 作为碳前体和氮源,依次通过静电纺丝、炭化和活化处理制得富氮分级多孔碳纳米纤维(N-PCNFs),用于室温下催化氧化H2S,其制备过程见图10(a)。
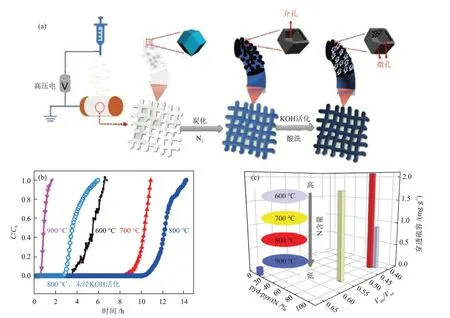
图10 N-PCNFs的制备过程示意(a)、不同炭化温度、KOH活化的N-PCNFs和炭化温度为800 ℃、未经KOH活化的N-PCNFs的穿透曲线(b)以及不同炭化温度、KOH活化的N-PCNFs的穿透硫容随pyd-pyr/tN和Vmic/Vtot的变化(c)[55]Fig.10 Schematic diagram of preparation process of N-PCNFs (a), H2S breakthrough curves of N-PCNFs derived from different carbonization temperatures with KOH activation and carbonized at 800 ℃ without KOH activation (b) and changes of saturated sulfur capacities of N-PCNFs derived from different carbonization temperatures with KOH activation with pyd-pyr/tN and Vmic/Vtot (c)[55]
作者发现,炭化温度对N-PCNFs的脱硫性能影响显著,即随着炭化温度的升高,脱硫性能表现出先增大后减小的趋势,当炭化温度为800 ℃时制得的N-PCNFs 的穿透硫容达到最大值(1840 mg/g),不同炭化温度下、由KOH 活化的N-PCNFs 和800 ℃炭化温度下、未经KOH 活化的N-PCNFs 的穿透曲线见图10(b)。不同炭化温度、KOH 活化的N-PCNFs 的穿透硫容随pyd-pyr/tN(吡啶-N、吡咯-N含量与总氮含量之比)和Vmic/Vtot(微孔体积与总孔体积之比)的变化见图10(c),由图10(c)可知,炭化温度会影响N 含量及N-PCNFs 的物理结构,进而影响N-PCNFs的穿透硫容。N为H2S解离提供活性位点,具有互穿通道的一维结构可以促进反应物的扩散以及产物的迁移,微孔有效地增加比表面积,提供了更多的活性位点,中孔为单质硫提供了更多的储存空间。SUN等[56]设计了一种Co 修饰的N 掺杂中空碳纳米纤维(Co-NHCFs),并用于室温下催化氧化H2S。实验结果表明,在25 ℃、含水量为80%的条件下,与N 掺杂中控碳纳米纤维(NHCFs)相比,Co-NHCFs 的脱硫性能显著提高,穿透硫容由620 mg/g 增加至1920~3970 mg/g,当Co 的负载量为6%时,Co-NHCFs 的穿透硫容达到最大值(3970 mg/g)。Co 促进了N 掺杂碳的能带重排,并使电子在碳平面上重新分布,提高了路易斯碱性,增强了活性氧物种的生成,进而对H2S 的催化氧化起到促进作用。
综上所述,多孔碳材料本身的脱硫性能有限,通过杂原子掺杂、碱溶液浸渍和金属氧化物负载等方法对多孔碳材料进行改性,可以显著提高其室温下催化氧化H2S 的性能。目前,多孔碳材料在工业应用方面存在一定的局限性,如脱硫产物堵塞孔道、活性组分的消耗造成催化剂失活或中毒,催化剂的循环利用性能仍是目前急需优化的主要技术问题,未来还需要开展进一步的研究。
3 结语与展望
基于多孔碳材料在室温下催化氧化H2S方面表现出的潜力,本文对其室温下催化氧化H2S 的机理进行了总结,并对活性炭、介孔炭、碳纳米管、石墨烯和碳纤维等多孔碳材料在室温下催化氧化H2S的研究进展进行了综述。尽管多孔碳材料在室温下催化氧化H2S 的研究取得了一定进展,但是进一步规模化生产和工业应用仍存在许多关键性问题需要解决:(1)研究人员对于多孔碳材料室温下催化氧化H2S 的活性位点尚无定论,目前大多数研究人员认为活性位点是缺陷或边缘碳原子,少数研究人员认为活性位点是掺杂的杂原子。因此需借助理论计算或其他先进的表征技术继续进行深入研究,如采用同步加速器X 射线粉末衍射结合高分辨率电子显微镜研究硫在纳米孔上的沉积过程和催化剂的结构演变,为多孔碳材料在催化氧化H2S 过程中的构效关系提供更基础的理解。(2)目前,多孔碳材料室温下催化氧化H2S的研究大多处于实验室阶段,研究人员对复杂工业烟气条件下的研究较少,动态环境(如原料气的组成、湿度和分压)变化对催化氧化H2S 的影响尚不清楚,这是实现工业应用面临的最大挑战。在研究环境因素对脱硫性能的影响时,应重点考虑多孔碳材料的稳定性及使用寿命。(3)目前,基于多孔碳材料的催化剂在使用一段时候后存在失活或中毒现象,在实际工业应用中,催化剂的再生非常重要,因此需要重点考虑开发操作简单、成本低廉、再生效果好,以及可实现硫的资源化利用的再生方法。
猜你喜欢
科学大观园(2023年17期)2023-08-30 05:16:14
化学工程师(2023年1期)2023-02-17 15:09:48
辽宁化工(2022年6期)2022-07-01 01:32:18
煤气与热力(2021年2期)2021-03-19 08:55:50
理化检验-化学分册(2020年12期)2021-01-26 00:41:38
装备维修技术(2020年4期)2020-11-23 11:37:13
中国果树(2020年2期)2020-07-25 02:14:28
冶金动力(2020年5期)2020-06-15 06:34:30
上海农业科技(2019年1期)2019-02-22 01:51:28
安全、健康和环境(2018年7期)2018-08-03 08:03:56
