
加权基因共表达网络筛选胃癌关键功能模块与预后相关基因
2023-10-25 10:15武寒徐磊王苗苗崔忠泽吴淑华
国际医药卫生导报 2023年20期
武寒 徐磊 王苗苗 崔忠泽 吴淑华
滨州医学院附属医院病理科,滨州 256600
胃癌是第5 种常见的恶性肿瘤,也是世界癌症病死率的第3 大原因[1]。2018 年,世界范围内有1 033 701 例新病例和782 685例死亡与胃癌相关,其中多数在诊断时已处于局部晚期阶段[2-3]。晚期胃癌的预后之所以较差,通常是因为早期诊断生物标志物的不足和缺少有效的治疗[4]。因此,胃癌的预防和控制已经成为紧急的公共卫生问题,迫切需要研究其发生发展的潜在机制,找出新的治疗和诊断靶点,最终提高患者的生存率。加权基因共表达网络分析法(weighted gene co-expression network analysis,WGCNA)于2005年提出,并在官方网站上提供R数据包,可用于构建加权基因共表达网络,检测基因模块,将基因模块与临床特征相关联,来鉴定模块内中心基因[5-7]。2023年1月至4月,在本研究中,笔者利用WGCNA 共表达网络、蛋白互作网络和生存预后,最终筛选出5 个预后相关基因,为进一步发现新的生物标志及胃癌诊断和治疗的潜在靶点提供的理论依据。
资料与方法
1.数据的获取与差异表达基因的确定
基因表达谱数据GSE65801(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65801)可从公开的基因表达综合数据库(Gene Expression Omnibus,GEO)中搜索并下载。GSE65801 是基于GPL14550 平台的表达谱,共包含64 个样本,其中32 个为组织黏膜样本,32 个为组织肿瘤样本。应用R 软件鉴定差异表达基因(differently expressed genes,DEGs),并以|log2 FC|≥2.0 和矫正后P<0.05 为差异有统计学意义。
2.WGCNA
使用“WGCNA”R 包构建胃癌和正常样本中所有基因的共表达网络。首先对RNASeq 数据进行过滤,降低异常值,软阈值(β)由函数pickSoftThreshold 确定,并将最佳的powers 值保存在sft$powerEstimate,使构建出的网络更符合无尺度网络标准。利用该加权相关系数,将相关矩阵变换为邻接矩阵,进而变换为拓扑重叠矩阵(topological overlap matrix,TOM)[8-9]。根据之前选定的软阈值,将全部基因划分为不同模块,其中每个模块至少包含50 个基因,并应用动态树剪切法将相关性小于0.25 的模块合并,将所有基因划分为数个模块。随机选择400个基因作为TOM 热图确定每个模块中遗传因子表示的相对独立性和模块之间的高度独立性。最后,使用WGCNA 算法计算由每个模块的基因和样本组成的模块特征基因(ME)的皮尔森相关系数和P值,选择相关系数最高的模块进行后续分析。
3.基因本体论分析(gene ontology,GO)[10]、京都基因和基因组百科全书富集分析(Kyoto Encyclopedia of Genes and Genomes,KEGG)[11]
通过构建维恩图(http://bioinformatics.psb.ugent.be/webtools/Venn/)筛选最显著共表达模块的DEGs。使用R 包“org.Hs.eg.db”将基因名转化为entrezlID,并使用R 包“clusterProfiler”“org.Hs.eg.db”“enrichplot”和“ggplot2”包进行GO、KEGG。KEGG 是一个从高通量实验技术生成的大规模分子数据中了解高级功能和生物系统的数据库资源,而GO是一个主要的生物信息学工具,用于注释和分析基因的生物过程(BPs)、分子功能(MFs)和细胞成分(CCs),结果以前10位BPs、MFs、CCs和KEGG为基础进行可视化分析。
4.蛋白质相互作用(PPI)分析及筛选预后相关基因
利用STRING 在线数据库(Version 11.0,http://string-db.org)在线将之前获得的基因构建成PPI 共表达网络(minimum required interaction score>0.4),然后输出PPI网络。 使用Cytoscape 软件(Version 3.8.2,http://www.cytoscape.org/)打开在STRING 构建的PPI 网络,使用“MCODE”包筛选出PPI 网络中的重要模块,以其中最重要模块的基因作为研究对象。在线分析网站GEPIA[12](http://gepia. cancer-pku. cn/)对最重要模块中的基因采用Kaplan-Meier法进行总生存(OS)和无病生存(DFS)分析,并以log-rank 法检验。从结果中筛选出P<0.05 的基因,这些差异表达的基因被认为有统计学意义,筛选出两种生存均有统计学意义的基因作为预后相关基因。
5.统计学处理
使用R 软件(v.3.6.3)进行统计分析,以P<0.05 为差异有统计学意义。
结果
1.胃癌组织和正常组织的DEGs
在GSE65801 数据集中获取到一个包含2 222 个DEGs的基因集(GSE65801_DIFF)。与正常组织相比,其中1 140 个基因在癌组织中表达下调,1 082 个基因表达上调(图1)。
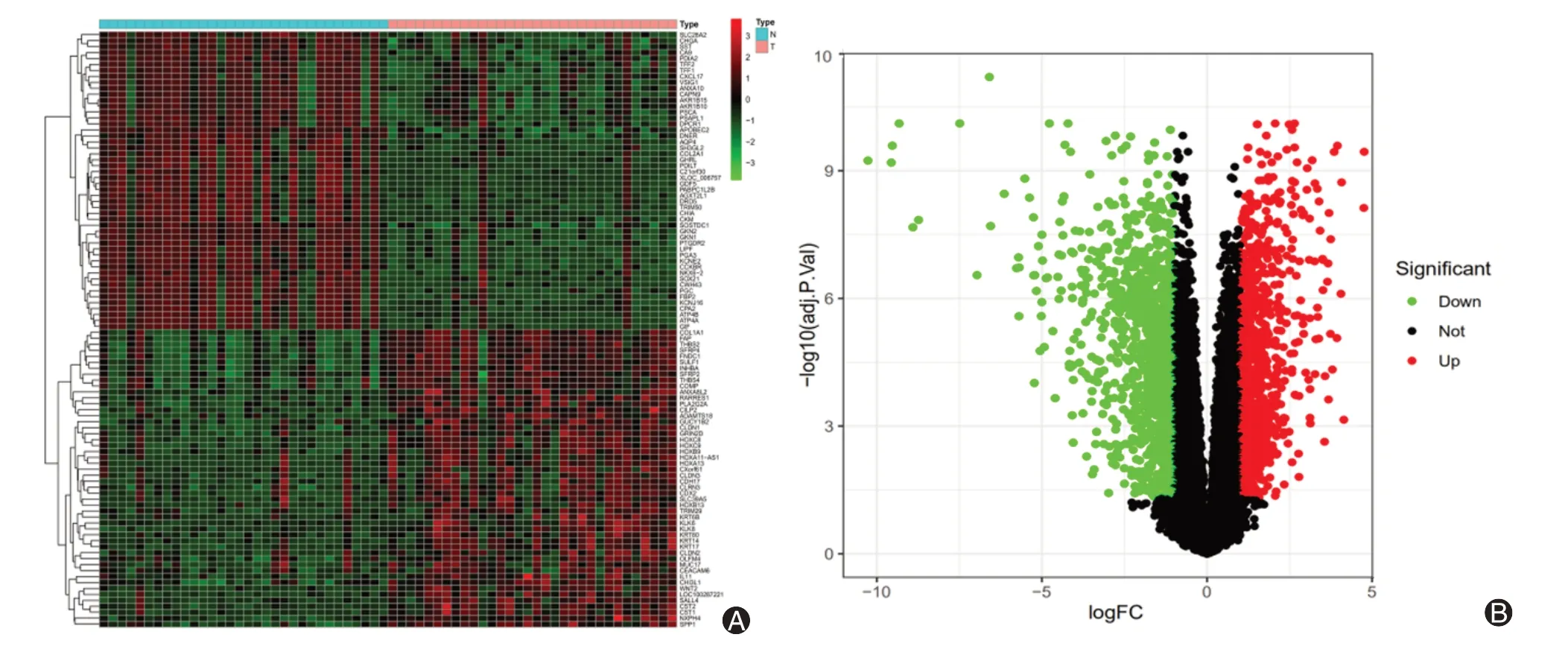
图1 胃癌和正常组织的差异表达基因。A:热图;B:火山图
2.对GSE65801数据集进行WGCNA的结果
从GSE65801数据集中获得核酸探针的矩阵,利用平台文件GPL14550将探针名转换为基因名,并对其基因表达量作标准化处理,最终得到的基因表达谱矩阵用于共表达网络构建。本研究将所有样本均纳入WGCNA。为了更符合无尺度特征,选取11 作为β 值构建共表达网络(图2A)。将剪切高度设置为0.25,合并模块,根据TOM 矩阵检测GSE65801,共发现12 个基因模块(图2B),随机选取400 个基因绘制TOM 热图,确定每个模块以及每个模块中基因独立存在(图2C)。从模块和性状的热图中可发现,淡青色(lightcyan)模块与GSE65801 中胃癌的相关性最高(图2D)。计算淡青色模块中基因MM 和GS 相关系数(cor=0.66,P=3.8e-115)以验证结果的可信度(图2E)。因此,选用GEO_lightcyan模块进行后续研究。
3.GO与KEGG富集分析
通过构建Venn 图(GEO_DIFF 和GEO_lightcyan)得到375个基因作为最显著模块的差异基因,这些基因作为关键基因进行后续分析(图3A)。使用R 软件富集分析结果显示,这些基因主要富集到糖胺聚糖代谢、血管发育、上皮细胞增殖、调节细胞对生长因子刺激的反应、调节细胞生长、细胞粘附等功能,以及PI3K-Akt、MAPK、Ras 等关键信号通路(图3B、C)。
4.关键基因筛选
构建了375 个关键基因的PPI 网络。使用Cytoscape 软件得到最重要模块有41 个基因(图4)。通过预后分析,获得了5 个预后相关基因:VCAN、SERPINE1、HGF、IGFBP7、FSTL3(图5)。

图4 对淡青色模块基因进行蛋白质相互作用分析(橙色区域为最重要功能模块,绿色为其他基因)
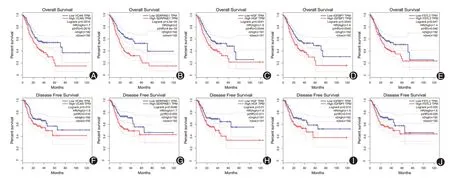
图5 GEPIA筛选预后相关基因。A、B、C、D、E:预后相关基因总生存分析;F、H、I、J:预后相关基因无病生存分析
讨论
胃癌是最常见的恶性肿瘤。近年来,虽然已有胃癌病因学的相关研究,但其发展的确切机制仍有待探索。本研究通过在GSE65801 数据集中进行WGCNA,获得了12 个共表达模块。选取出其中差异最显著的共表达模块进一步筛选差异基因,最终得到375个关键基因。
通过富集分析,笔者发现关键基因主要富集到糖胺聚糖代谢、血管发育、上皮细胞增殖、调节细胞对生长因子刺激的反应、调节细胞生长、细胞粘附等功能上,除此以外,还富集在PI3K-Akt、MAPK、Ras 等关键信号通路上。已有研究表明,在胃癌中,糖胺聚糖可通过多种途径影响肿瘤细胞的生物学行为,从而影响患者预后[13]。血管发育是肿瘤细胞生长的必要条件之一[14],而细胞的增殖与生长是促进肿瘤进展不可或缺的因素[15],二者可相互作用影响胃癌的进展。众多学者也已证明,PI3K-Akt[16-17]、MAPK[18-19]、Ras[20]等信号通路对研究胃癌发生机制具有重要意义。除此之外,笔者还发现这些基因亦富集到许多新的功能与通路上,这可能为进一步探究胃癌发生发展规律、探索胃癌治疗方法、改善患者生存提供新思路。
本研究将关键基因进行PPI分析,并将最重要模块中的基因进行预后分析,最终鉴定出5 个预后相关基因:VCAN、SERPINE1、HGF、IGFBP7、FSTL3。通过在线网站GeneCsrds(https://www.genecards.org/)检索可知,VCAN 又被称为Versican,可能参与细胞运动、调节细胞生长和分化;SERPINE1作为尿激酶型纤溶酶原激活物抑制剂之一,可参与细胞粘附和扩散的调节;HGF 可调节多种组织类型中的细胞生长、细胞运动性和形态的发生;IGFBP7 是胰岛素超家族的生长促进肽成员,可与胰岛素结合对细胞进行调控;FSTL3是编码卵泡抑素模块蛋白家族的一种分泌糖蛋白。
Li 等[21]研究发现,VCAN 是影响胃癌预后的独立危险因素。有研究表明,VCAN 的表达在胃癌中显著增高,并与肿瘤的生长和发展密切相关[22]。在本研究中,VCAN 被富集到粘多醣代谢过程上,提示其可通过调节癌细胞生长分化促进胃癌进展。Xu 等[23]研究证实,SERPINE1 可通过调节葡萄糖代谢来促进乳腺癌的发生。研究发现,分泌型SERPINE1 表达增加,可诱导基质金属蛋白酶1(MMP1)的表达并使结肠癌转移风险提高[24]。本研究发现,SERPINE1 被富集到血管生成相关通路上,这与Teng 等[25]研究结果一致,该研究发现SERPINE1 表达上调和激活VEGFR-2 信号通路最终促进肿瘤血管生成,影响胃癌进展。Gao 等[26]研究发现,HGF 表达可能与胃癌中血管生成和淋巴结转移呈正相关。Ding 等[27]进一步证实,肿瘤相关成纤维细胞的HGF可通过PI3K/AKT和ERK1/2信号促进胃癌血管化。本研究结果显示,HGF被富集到血管发生、间充质细胞分化等重要功能以及PI3K-Akt 信号通路、MAPK 信号通路等通路上,说明HGF对肿瘤细胞生长发育、信号传导等过程具有重要意义。最新研究发现,IGFBP7 可作为膀胱癌的治疗靶点[28]。IGFBP7 与肝细胞癌[29]、急性淋巴细胞白血病[30]、胃癌[31]等疾病均密切相关。肿瘤相关成纤维细胞分泌的IGFBP7 通过FGF2/FGFR1/PI3K/AKT 轴增强肿瘤相关巨噬细胞的浸润,从而促进胃癌的发生[32]。本研究结果显示,IGFBP7 被富集到细胞生长相关的功能上,表明调控IGFBP7有望成为胃癌调控的重要靶点。FSTL3在胃癌组织中高表达,并与总体无病生存期相关[33]。本研究结果显示,FSTL3 被富集到跨膜受体蛋白丝氨酸/苏氨酸激酶信号通路、细胞粘附的正调控、调节细胞对生长因子刺激的反应等功能上,说明其可通过多种途径影响胃癌的发生。
综上所述,本研究对GSE65801 数据集进行分析,发现了胃癌发生发展过程中重要的功能模块,并在胃癌关键功能模块中筛选出5 个可作为胃癌预后生物标志物或潜在治疗靶点的预后相关基因:VCAN、SERPINE1、HGF、IGFBP7、FSTL3。这些基因及其所富集到的通路及功能可能为临床研究提供新方向。然而,本文只是从生信水平进行了初步分析,由于样本量的限制、数据集来源存在一定的局限性,仍需扩大样本量分析并从多层面开展分子机制、细胞实验、动物实验等更深入的探究,进行进一步验证。
利益冲突所有作者均声明不存在利益冲突
作者贡献声明武寒:酝酿和设计试验,实施研究,采集数据,分析/解释数据,起草文章,对文章的知识性内容作批评性审阅,统计分析;徐磊:酝酿和设计试验,实施研究,分析/解释数据,起草文章,对文章的知识性内容作批评性审阅,统计分析;王苗苗:酝酿和设计实验,实施研究,采集数据,对文章的知识性内容作批评性审阅,统计分析;崔忠泽:酝酿和设计试验,实施研究,分析/解释数据,对文章的知识性内容作批评性审阅,统计分析;吴淑华:酝酿和设计实验,分析/解释数据,对文章的知识性内容作批评性审阅,统计分析获取研究经费,行政、技术或材料支持,指导,支持性贡献
猜你喜欢
昆明医科大学学报(2020年12期)2021-01-26
山东医药(2017年20期)2017-07-01
中国学术期刊文摘(2016年1期)2016-02-13
中国卫生标准管理(2015年3期)2016-01-14
中国病理生理杂志(2015年8期)2015-12-21
医学研究杂志(2015年9期)2015-07-01
医学研究杂志(2015年3期)2015-06-10
中国当代医药(2015年20期)2015-03-01
山东医药(2015年44期)2015-02-28
创业家(2015年1期)2015-02-27
