
新生儿精氨酰琥珀酸尿症一例并文献复习
2023-09-23 12:05胡启发高镝苏喆
海南医学 2023年17期
胡启发,高镝,苏喆,3
1.遵义医科大学珠海校区,广东 珠海 519041;
2.深圳市儿童医院新生儿科,广东 深圳 518026;
3.深圳市儿童医院内分泌科,广东 深圳 518038
精氨精氨酰琥珀酸尿症(argininosuccinic aciduria,ASA)又称精氨酰琥珀酸裂解酶缺乏症(argininosuccinic acid lyase deficiency,ASLD),是尿素循环障碍性疾病的一种类型,属于一种罕见的常染色体隐性遗传病。据估计,国际上ASLD的发病率约为1∶218 750。ASLD 根据患儿临床表现可分为早发型和晚发型,早发型较为多见[1]。ASLD的临床表现复杂多变,患儿既可以无临床表现,也可以出现呕吐、拒乳、嗜睡、黄疸、惊厥、肝脏肿大、肌张力低下和高氨血症的相关症状,包括肝功能障碍、神经认知缺陷、行为异常和学习障碍等。新生儿期发病的患儿极为凶险,可出现严重的高氨血症,甚至危及生命。本病在国内外的报道相对较少,本文将对1 例ASLD 新生儿的临床资料及家系全外显基因检测结果进行分析,总结ASLD 的临床特点及基因突变情况。
1 病例简介
因“反应差半天”为主诉于2021年12月25 日就诊于深圳市儿童医院新生儿科,患儿系G2P2,男,出生3 d,40+1周顺产出生,产时羊水Ⅰ度污染,无胎膜早破、胎盘早剥及窒息史,出生体质量3 040 g,人工喂养,进乳减少,难以唤醒,刺激足底可睁眼,无哭声,伴自主活动明显减少;母亲38岁,有妊娠期糖尿病及妊娠期甲状腺功能减退症,予饮食调节血糖、定期口服优甲乐治疗,否认妊娠期高血压,否认毒物及放射线接触史,产前无发热,无使用抗生素、镇静剂及地塞米松;父亲39岁,体健,非近亲结婚,无家族遗传病史;哥哥6 岁,体健,出生史无特殊,无类似症状。入院查体:体温36.7℃,心率158 次/min,呼吸52 次/min,血压45/39 mmHg(1 mmHg=0.133 kPa),体质量2.84 kg,浅昏迷状态,反应差,无哭声,全身皮肤轻度黄染,皮肤弹性一般,双侧瞳孔对光反射尚灵敏,四肢肌张力增高,偶有一过性肢体抖动。实验室检查,血常规:白细胞22.98×109/L,淋巴细胞12.68×109/L,中性粒细胞7.88×109/L,红细胞4.51×1012/L,血红蛋白127 g,血小板669×109/L;血气分析:pH 7.302,二氧化碳分压(PCO2):29.1 mmHg,氧分压(PO2):24 mmHg,碱剩余(BE):-12 mmol/L;生化检测:血钠139.0 mmol/L,血钾2.52 mmol/L,乳酸7.40 mmol/L,谷氨酸转氨酶667 U/L,丙氨酸转氨酶180 U/L,天冬氨酸转氨酶246 U/L,总胆红素406.5 μmol/L,直接胆红素237.4 μmol/L,间接胆红素169.1 μmol/L,总蛋白36.7g/L,白蛋白25.0 g/L;凝血功能:活化部分凝血活酶时间(APTT)85.1 s,凝血酶原时间(PT)32 s,国际标准化比值(INR)3.05 inr,凝血酶时间(TT)23.54 s,纤维蛋白原(FIB)0.6 g/L;血氨(入院时)468.6 μmol/L,血氨(峰值)578.5 μmol/L;颅脑MR平扫(3.0T)+脑功能成像(MRS)(患儿1个月22 d时):左侧顶叶及部分枕叶脑软化灶形成;MRS提示神经元受损伤并胶质增生。新生儿神经行为评估(NBNA):20分。
因患儿血氨明显升高,考虑遗传代谢相关性疾病,行血液串联质谱检测显示瓜氨酸182.96 μmol/L,尿有机酸分析显示乳清酸10.4 μmol/L,4-羟基苯乳酸67.7μmol/L,考虑尿素循环障碍性疾病。经家长同意,并签署相关知情同意书后,抽取患儿及其家系(包括同胞哥哥以及父母)的外周血液,委托广州嘉检医学检验中心进行全外显基因测序,基因报告示患儿ASL基因复合杂合突变,一方面,存在来自父亲的c.925G>A(p.G309R)错义突变,该突变位点的染色体位于chr7:65554269,其中第925 位的碱基鸟嘌呤错义突变为腺嘌呤,该突变位点位于既往报道的染色体位置,致病性分析为可能致病性变异;同时,存在来源于母亲的c.1251-1G>A 错义突变,该突变位点的染色体位于chr7:65557754,其中第1251-1位的碱基鸟嘌呤错义突变位腺嘌呤,致病性分析为可能致病性变异。父母双方基因突变位点均属于杂合变异。父母表型均为正常;同胞哥哥,6岁,同样存在ASL基因变异c.925G>A(p.G309R),但无ASL基因c.1251-G>A的变异,且表型正常,见图1。该患儿诊断为:(1)精氨酰琥珀酸尿症(ASL_NM000048.4 基因变异);(2)新生儿休克;(3)呼吸衰竭;(4)弥漫性血管内凝血;(5)胆汁淤积性肝病;(6)呼吸性碱中毒;(7)电解质紊乱(低钾血症、低钙血症);(8)低蛋白血症;(9)肝功能损害;(10)血小板增多症。本例患儿经血液透析、补充精氨酸等降血氨、左卡尼汀改善代谢、维生素B6营养神经、呼吸机辅助通气、高压氧治疗促进脑损伤恢复及低蛋白、高热量饮食支持等综合治疗后,血氨由578.5 μmol/L 逐渐降低至53.2 μmol/L,进乳、自主活动及精神状态较前好转。患儿住院治疗50+d,病情好转后出院,出院后继续予低蛋白、高热量饮食,补充精氨酸等治疗,出院12 d 后复查血氨82.7 μmol/L。目前患儿为生后3 个月,精神反应及自主活动良好,无抽搐及喂养困难。
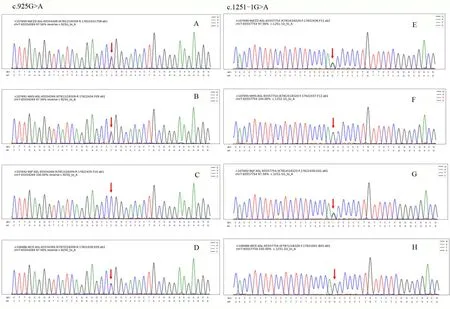
图1 患儿、父母及同胞哥哥ASL基因突变位点c.925G>A、c.1251-1G>A验证测序图Figure 1 Sequence diagram of the ASL gene mutation site c.925G>A,c.1251-1G>A of the patient,parents,and his brother
2 讨论
尿素循环是氨解毒的主要途径。尿素循环障碍性疾病是一种因尿素循环过程中所需的酶或转运蛋白的缺陷,导致氨解毒和精氨酸合成障碍的遗传代谢性疾病。ASLD是仅次于鸟氨酸氨甲酰转移酶缺乏症(OTCD)的第二常见的尿素循环障碍性疾病,约占尿素循环障碍性疾病的16%[2]。精氨酰琥珀酸裂解酶(ASL)是由ASL 基因编码产生的一类酶,位于细胞质中,主要在肝脏、肾脏、小肠以及脑中表达。该酶是尿素循环过程中将氨转化为尿素的6 种酶之一,参与尿素循环的第4步生化反应,通过催化ASA产生精氨酸和富马酸。ASL 基因的突变导致精氨酰琥珀酸裂解酶缺乏,从而引起ASA 的切割障碍,使得ASA 的积累、血浆中瓜氨酸及精氨酸琥珀酸水平升高、尿液中的精氨酸琥珀酸过度排泄[3-4]。
人体内的血氨可以通过自由扩散的形式通过血脑屏障,并且通过星形胶质细胞内的谷氨酰胺合成酶与谷氨酸盐快速浓缩,从而产生具有渗透活性的谷氨酰胺。此外,氨本身可以扰乱血钾的稳态,并影响水经水通道蛋白的转运,引起星形胶质细胞肿胀,并导致细胞毒性脑水肿[5]。一旦人体出现代谢危机,大量的氨就会在血液及大脑中聚集,导致神经功能障碍,出现震颤、共济失调、癫痫发作、昏迷,甚至死亡。正常情况下,早产儿血氨水平<150 μmol/L,足月儿<100 μmol/L[6]。在大多数新生儿患者中,高氨血症是尿素循环障碍的重要标志,其血氨浓度峰值>500 μmol/L。血氨浓度正常的患儿几乎可以排除有症状新生儿的尿素循环障碍。当血氨浓度超过200 μmol/L时,患儿可出现嗜睡、浅昏迷等神经系统症状,严重者可致死亡[6-7]。1957 年第一次报道1 例ASLD病例,自此之后,国外陆续报道了有关ASLD的一些病例,并进行了相关的研究[8]。2014年至今,国内报道新生儿精氨酰琥珀酸裂解酶(ASL)基因变异的病例共有5 例,大多以反应差、喂养困难就诊,并且血氨水平均明显增高,最终经基因分析确诊为精氨酰琥珀酸裂解酶缺乏症(即精氨酰琥珀酸尿症)[9-13]。本例患儿生后3 d,以反应差来我院就诊,四肢肌张力增高,偶有一过性肢体抖动,血氨水平(入院时)468.6 μmol/L,血氨(峰值)578.5 μmol/L,血液串联质谱检测提示血浆瓜氨酸水平显著升高,尿有机酸分析显提示尿液中乳清酸及4-羟基苯乳酸水平升高,符合尿素循环障碍性疾病的临床特点,因此,入院后初步考虑为尿素循环障碍。经患儿家长同意并签署相关知情同意书后,予行家系全外显基因测序,基因报告显示患儿存在与ASLD 表型相关的可能致病性变异,分别来自父亲和母亲的c.925G>A(p.G309R)和c.1251-1G>A 错义突变,根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)指南,该患儿相关基因突变的致病性分析为可能致病性变异。其中,c.925G>A 突变在国外已有相关报道[14],但在国内目前仍鲜有文献报道。
ASLD 的临床表现缺乏特异性,对于该病诊断主要依靠生化检测和基因分析,血氨水平、血串联质谱检测及尿有机酸分析有助ASLD 于早期识别ASLD,但明确诊断仍有赖于基因检测分析[15]。本例患儿父母及同胞哥哥均存在ASL 基因杂合突变,但父母双方及同胞哥哥均表型正常,患儿存在来源于父母双方的ASL 基因复合杂合突变,遗传累积效应致病,符合该病常染色体隐性遗传的形式,结合血液串联质谱检测以及尿有机酸检测分析结果,明确诊断为ASLD。
ASLD 的治疗可分为急性期和慢性期的治疗;急性期的治疗是在代谢失代偿期间快速控制高氨血症,慢性期的治疗主要包括限制蛋白质摄入和补充精氨酸。饮食的管理是ASLD 患儿长期管理的主要支柱。在长期管理中,尽管予低蛋白饮食、补充精氨酸治疗,但部分患儿仍有代谢失代偿或血氨升高,这时可予口服氮清除治疗。本例患儿经控制饮食、营养支持、降血氨、补充精氨酸等综合治疗,血氨由578.5μmol/L 降低至53.2 μmol/L,进乳及精神反应较前好转。出院后,患儿继续低蛋白、高热量饮食,补充精氨酸等治疗,出院12 d 后复查血氨82.7 μmol/L,目前患儿为生后3个月,精神反应及自主活动良好,无抽搐及喂养困难。
近年来,肝移植被推荐用于治疗各种代谢性疾病,包括尿素循环障碍性疾病。在尿素循环障碍性疾病的患者中,肝移植的目标是最大限度地降低复发性高氨血症危象和进行性神经损伤的风险。据报道,对于尿素循环障碍性疾病的患儿,接受肝移植后5 年和10 年的生存率均接近90%。但是,尿素循环障碍性疾病的患者进行肝移植的标准仍不清楚。另外,器官捐赠也是一个重要问题。器官捐赠的标准非常严格,很难为儿科患者找到匹配的良好的成人供体[16-17]。除此之外,还有肝细胞移植以及基因治疗,但该技术均未成熟,目前仍处于试验摸索阶段。
综上所述,临床上有神经系统异常表现、喂养困难、血氨显著升高的患儿,需警惕尿素循环障碍性疾病,行血串联质谱检测和尿有机酸分析有助于临床诊断,尽早行基因检测分析对该病的明确诊断和预后有重要意义。该病预后不良,再生育应进行遗传学指导。
猜你喜欢
中国民间疗法(2021年18期)2021-11-02
首都食品与医药(2021年5期)2021-03-22
浙江工业大学学报(2017年5期)2018-01-22
转化医学电子杂志(2018年11期)2018-01-16
国外医药(抗生素分册)(2016年3期)2016-07-12
中国现代药物应用(2016年10期)2016-03-06
中外医疗(2015年11期)2016-01-04
中国当代医药(2015年30期)2015-03-01
中国洗涤用品工业(2015年9期)2015-02-28
中国实用医药(2013年14期)2013-10-19
