
应用CRISPR/Cas9技术建立ERK激酶相分离荧光探针定点整合的稳定细胞株
2023-09-01 10:41:40杨玉梅张坤晓
生物技术通报 2023年8期
杨玉梅 张坤晓
(1. 江苏海洋大学药学院,连云港 222005;2. 江苏海洋大学江苏省海洋药物活性分子筛选重点实验室,连云港 222005)
基于液-液相变原理(liquid-liquid phase separation)开发的激酶荧光探针(separation of phasebased activity reporter of kinase, SPARK)是近年来继分离荧光蛋白技术(split fluorescent protein, split FP)、循环排列的荧光蛋白技术(circularly permuted fluorescent protein, cpFP)以及荧光共振能量转移技术(fluorescence resonance energy transfer, FRET)之后的第4种研究蛋白-蛋白相互作用(protein-protein interaction, PPI)的荧光探针技术[1-11]。该技术的原理是利用一对同型寡聚体小肽(四聚体小肽命名为HOTag6,六聚体小肽命名为HOTag3)的多价结合和迅速扩增,将瞬时的PPI转变成稳定的绿色荧光的形态变化,表现为均匀分布的GFP绿色荧光聚集成高亮度的绿色液珠[12-15]。以检测ERK激酶活性的ERK-SPARK探针为例,具体做法是将来源于细胞周期蛋白Cdc25C的一段可被ERK识别并被ERK磷酸化的肽段序列(未被磷酸化的肽段记为Perk,磷酸化后的肽段记为Perk-Phospho)与HOTag3(6聚体)融合,而将可识别、结合磷酸化的该肽段的WW结构域与HOTag6(4聚体)融合,并在中间插入荧光蛋白EGFP来获取荧光信号[16]。当用EGF处理表达ERK-SPARK探针的细胞时,Perk被ERK磷酸化为Perk-Phospho,之后WW结构域识别并结合到Perk-Phospho上。由于六聚体和四聚体的不对称性,6×Perk-Phospho和4×WW结合后会空余出2×Perk-Phospho,这个2×Perk-Phospho又会结合4×WW,此时会空余出2×WW,如此循环往复,两组分越聚越多,在极短的时间内即可形成明显的绿色液滴,聚集的荧光信号是单分子GFP亮度的30-50倍,具有很高的辨识度(图1)。这类探针可以极高的分辨率和灵敏度在活细胞内实时观测激酶活性的动态变化,因此理论上有较大的应用前景,特别是在蛋白激酶抑制剂高通量筛选平台的建立上。
但在实践中发现,SPARK探针仅能以瞬时表达的方式在细胞内发挥功能,当采用病毒载体将编码SPARK探针的序列整合至宿主染色体上时,细胞内一种尚不明确的机制会导致SPARK探针的功能完全丧失,因而限制了该技术的应用。且SPARK探针功能的发挥蛋白表达量有密切关系,瞬时表达的不稳定性影响实验结果的准确性。CRISPR/Cas9 技术是利用Cas9 核酸酶与sgRNA组成的复合体对特异性靶点识别,在基因组上的特定位置对DNA进行双链剪切。发生双链基因组断裂的DNA可以通过真核细胞自然存在的非同源末端结合(NHEJ)或同源重组(HR)修复,其中同源重组修复机制可将外源片段整合到基因组指定位点,因此CRISPR/Cas9基因组编辑技术可以在基因组层面实现定点基因敲入的操作,且该技术具有效率高、研发周期短等特点。
为了克服使用病毒载体对SPARK探针的潜在影响,本研究利用CRISPR/Cas9介导的同源重组技术[17-19],在不使用任何病毒相关载体的条件下,将ERK-SPARK荧光探针基因表达盒定点插入人AAVS1位点(位于人PPP1R12C基因第一个内含子内,外源基因插入此位点可长期正常转录且不影响细胞其他基因)[20],成功构建了可被多西环素(doxycycline)诱导表达ERK-SPARK荧光探针的稳定KYSE-150细胞株。该稳定细胞株解决了SPARK荧光探针瞬时表达方式表达量不可控问题,将SPARK探针的序列定点整合至宿主染色体上,可使其长期正常转录且不影响细胞其他基因。为后续将SPARK技术应用于高通量筛选蛋白激酶抑制剂奠定了前期基础。
1 材料与方法
1.1 材料
人食管鳞癌细胞株(KYSE-150)购于上海北诺生物科技有限公司。pX330、pUC19、pCW-Cas9、pcDNA3.1-ERK-SPARK质粒购于Addgene。Bbs I酶、BsmB I酶、T4 DNA连接酶购自 New England Biolabs(NEB)公司。质粒抽提试剂盒购自Qiagen公司。DNA提取试剂盒、In-Fusion无缝克隆试剂盒购自TaKaRa公司。Lipofectamine 2000试剂、高糖DMEM培养基、胎牛血清和胰酶均购自Gibco公司。兔抗GFP的单抗购自美国CST公司。辣根过氧化酶标记的山羊抗鼠兔IgG购自北京中杉金桥生物技术有限公司。多四环素购自陶素化学。寡核苷酸序列由北京擎科公司合成。
1.2 方法
1.2.1 sgRNA寡核苷酸链合成 使用CRISPR在线设计工具(https://portals.broadinstitute.org/gppx/crispick/public)根据评分系统,设计了3个20 bp的sgRNA(sp1、sp2 和 sp3)。分别在编码链模板 5' 端添加 CACCG,非编码链模板 5' 端添加AAAC,3' 端添加C。由北京擎科公司合成。设计的寡核苷酸序列见表1。

表1 构建 sgRNA 表达载体引物、基因组扩增引物信息Table 1 PC construction of sgRNA expression vector primers and genome amplification primer information
1.2.2 CRISPR/Cas9 打靶载体构建 用 T4 DNA 连接酶将Oligo退火形成的双链二聚体与 Bbs I酶切的pX330 骨架质粒连接,转化DH5α 感受态细胞,均匀涂布于 Ampicillin 抗性的 LB琼脂培养板上。挑取单菌落进行测序鉴定,扩增鉴定连接成功的菌液,并用质粒抽提试剂盒提取。
1.2.3 AAVS1位点同源重组供体质粒的设计与构建 供体质粒设计的示意图如下(图2)。供体质粒两端750 bp的同源臂通过150基因组PCR获得;Tet-On 3G表达系统、嘌呤霉素抗性基因序列克隆自pCW-Cas9质粒;ERK-SPARK基因序列及突变体阴性对照克隆自pcDNA3.1-ERK-SPARK质粒。所获得的片段使用In-Fusion无缝克隆按图2中的顺序连接。

图2 ERK-SPARK knock-in质粒设计示意图Fig. 2 Design schematic diagram of ERK-SPARK knock-in plasmid
1.2.4 细胞转染 将150细胞接种于6孔板中,待其密度接近90%时,每孔分别转入1 μg CRISPR/Cas9 打靶载体与1 μg的供体质粒。转染48 h后,将细胞分别转入10 cm的皿中继续培养两周,之后加入2 μg/mL的嘌呤霉素,筛选出对嘌呤霉素具有抗性的细胞亚群。
1.2.5 流式细胞分选 在获得对嘌呤霉素具有抗性的细胞亚群后,提前24 h加入2 μg/mL的dox,之后将细胞消化进行流式细胞分选,收集GFP阳性细胞。
1.2.6 ERK-SPARK蛋白水平鉴定 将获得的同时对嘌呤霉素具有抗性、且GFP表达阳性的细胞提取总蛋白,经SDS-PAGE电泳并转至PVDF膜,用5%脱脂奶粉封闭1 h,之后GFP一抗4℃孵育过夜,1×TBST 清洗后,二抗溶液室温孵育1 h,1×TBST再次清洗后,用化学发光仪检测ERK-SPARK荧光探针蛋白表达情况。
1.2.7 ERK-SPARK荧光探针成像实验 将获得的重组细胞铺接种在96孔板中。加入终浓度为100 ng/mL终浓度的EGF,孵育10 min后置于荧光显微镜下观察。
2 结果
2.1 同源重组所构质粒的检测结果
本研究中所构建的质粒经北京擎科公司测序检测后,序列与设计图谱完全一致。
2.2 构建AAVS1位点敲入的ERK-SPARK稳定细胞株
若ERK-SPARK基因表达盒成功插入AAVS1位点后,重组细胞将获得以下性状:(1)对嘌呤霉素的抗性,这是因为嘌呤霉素基因自身不带启动子,由PPP1R12C基因的启动子启动表达,因此只有正确插入AAVS1位点的细胞才会获得对嘌呤霉素的抗性;(2)在加入dox后可检测到GFP荧光信号。这种设计的优势在于避免了单克隆细胞株的筛选,大大缩短了稳定细胞系的构建。用嘌呤霉素和流式细胞双重筛选的策略,排除了部分没有发生基因重组、但被周围具有嘌呤霉素抗性细胞所保护的假阳性细胞。对所获得的重组细胞提取基因组DNA、并进行PCR鉴定后发现扩增得到的片段大小约为6200 bp,与插入片段的大小一致(图3)。Western blot结果(图4)显示,在加入dox诱导24 h,可以检测到ERKSPARK融合蛋白的条带,大小分别为51 kD与42.4 kD,与实际大小相符。荧光成像实验(图5)显示,加入dox WT诱导24 h,之后加入EGF激活ERK信号通路后,可观察到ERK-SPARK的相变信号,而突变体对照组则无信号,这表明本研究所采用的构建SPARK稳定细胞系的方法不影响SPARK的功能。

图3 基因组PCR检测KYSE-150细胞株AAVS1位点ERK-SPARK表达Fig. 3 Genomic PCR detection of the ERK-SPARK expression at the AAVS1 locus in KYSE-150 cells

图4 Western blot 验证ERK-SPARK在 KYSE-150细胞株中诱导表达Fig. 4 Validating the doxycycline-induced expression of the ERK-SPARK reporter in KYSE-150 cells by Western blot
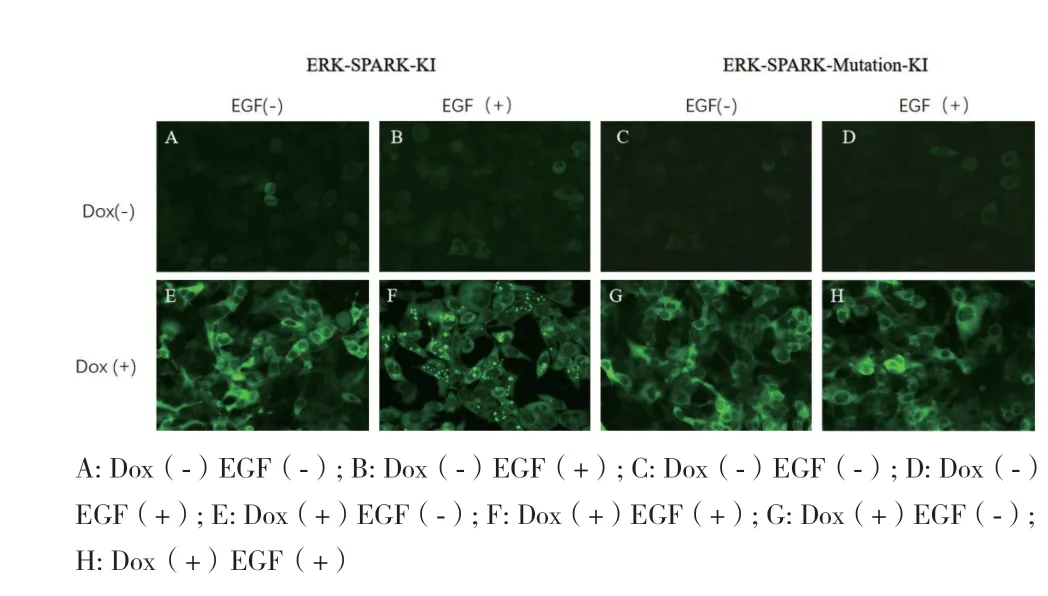
图5 EGF处理后 ERK-SPARK knock-in细胞系荧光成像(20X)Fig. 5 Fluorescence images of ERK-SPARK knock-in cells stimulated with EGF(20X)
3 讨论
3.1 SPARK探针具有反应灵敏迅速等优点,应用前景广阔
PPI介导了细胞信号转导通路,其紊乱与心血管疾病、癌症等重大疾病的发生发展密切相关,因此开发检测PPI的荧光探针一直是生物技术领域重要的前沿方向之一。通过设计特定的荧光探针检测蛋白激酶与其底物的相互作用、从而研究细胞内的“磷酸化事件”又是这类荧光探针设计开发的热点。在SPARK技术出现之前,荧光能量共振转移技术(FRET)是较为常用的研究蛋白磷酸化修饰的工具。但FRET技术的信号强度弱、信噪比不高的缺点限制了其在活体成像及高通量筛选上的应用。基于相分离原理开发的SPARK技术可以实时地捕获特定靶点瞬间的磷酸化事件,并通过将单分子荧光蛋白快速聚集成直径为1-2 μm大小的液滴的方式,将磷酸化信号转换为荧光信号,从而以直观的方式对激酶活性进行可视化的检测和定量,是一种全新的、可实时监测激酶活性的工具。
3.2 SPARK瞬时表达的使用方式具有弊端
尽管SPARK技术较FRET技术有诸多的优点,但SPARK技术同样存在一些明显的缺陷。首先,由于SPARK技术的设计原理是基于液-液相分离,因此SPARK探针需要在细胞内维持较高的表达水平才能发挥其功能,所以在构建SPARK探针的质粒时需采用强启动子启动SPARK的基因表达盒、且需要有较高的细胞内拷贝数。其次,在实践中发现,采用任何形式的病毒载体将SPARK探针整合到宿主细胞基因组上时都将导致SPARK探针的失效,无法像FRET技术那样通过慢病毒感染的方式构建表达SPARK探针的稳定细胞系,因此只能通过瞬转的方式将SPARK送入细胞,在具体的应用场景中会导致细胞的不均一性,部分细胞的激酶活性将无法被检测到,且当应用在激酶抑制剂的高通量筛选时会增加筛选的步骤与复杂性。最后,在设计任一激酶的SPARK探针时,需要慎重选择、测试、优化可被该激酶磷酸化的特异性肽段序列,以及可识别、结合磷酸化后的肽段的结构域;若二者的亲和力过高,则极易导致假阳性的出现,这是导致部分激酶无法设计为SPARK荧光探针的重要原因。
3.3 CRISPR/Cas9基因编辑技术使SPARK荧光探针的使用更加稳定可靠
本研究构建的AAVS1位点插入的诱导型ERKSPARK稳定细胞系规避了SPARK技术所存在的部分缺陷,通过采用多四环素诱导的Tet-On 3G系统实现了SPARK探针以可控的方式在细胞内的高表达,而不使用病毒载体的方式保证了SPARK探针在细胞内功能的有效性。利用该稳定细胞株对ERK激酶抑制剂进行筛选,可以将小分子药物的影响可视化,结合可视化高通量筛选仪器,以荧光信号为标准,对细胞质液滴成像情况进行统计,可提高激酶抑制剂的筛选效率。后续,所构建的稳定细胞系将被应用于ERK激酶抑制剂的高通量筛及探索ERK信号通路中关键调控机制的相关研究中。
4 结论
本研究利用CRISPR/Cas9介导了同源重组技术,将ERK-SPARK荧光探针定点整合到了人AAVS1位点中,成功构建了诱导表达型的SPARK荧光探针的稳定细胞系。解决了SPARK探针无法通过慢病毒等方式构建为稳定细胞系、进而限制该技术应用场景的瓶颈问题。
猜你喜欢
天津医科大学学报(2021年3期)2021-07-21 09:03:46
世界科学技术-中医药现代化(2021年12期)2021-04-19 12:31:40
天津医科大学学报(2019年6期)2019-08-13 07:04:42
中国医药生物技术(2015年4期)2015-12-26 08:26:36
安徽医科大学学报(2015年9期)2015-12-16 11:09:42
医学研究杂志(2015年11期)2015-06-10 06:44:03
中国当代医药(2015年16期)2015-03-01 02:03:11
中国医药导报(2015年27期)2015-02-28 22:08:02
癌变·畸变·突变(2014年2期)2014-03-01 04:39:42
遗传(2014年3期)2014-02-28 20:59:01
