
普克鲁胺中间体3-氟-4-异硫氰酸根-2-三氟甲基苯甲腈的合成
2023-08-19 00:30:04刘双双詹乐武李斌栋
合成化学 2023年8期
刘双双, 侯 静, 王 娟, 詹乐武, 李斌栋
(南京理工大学 化学与化工学院,江苏 南京 210094)
普克鲁胺(Proxalutamide,图1)作为新一代雄激素受体(AR)拮抗剂,对转移性前列腺癌和转移性乳腺癌具有良好的治疗效果[1-4]。 Proxalutamide是抗前列腺癌新药恩杂鲁胺(Enzalutamide, MDV3100)的核心结构,是采用靶蛋白晶体结构的计算机辅助设计并经反复优化后得到的新型化合物,与恩杂鲁胺(MDV3100)相比,其化学结构存在多处改变,从而导致性质产生了较大变化,例如Proxalutamide改善了分子溶解度和动力学性质[5-6]。自2020年新冠疫情爆发后,科学家将Proxalutamide的治疗领域应用到新冠病毒方面,研究发现,Proxalutamide对治疗新冠轻症和中症患者效果显著,目前仍处于临床阶段[7]。
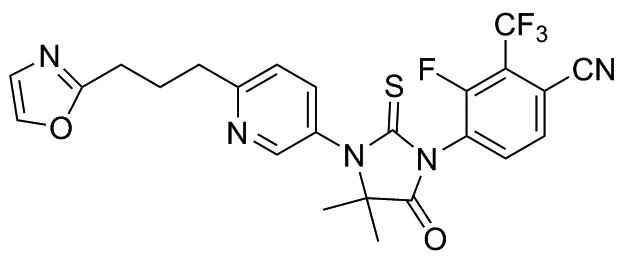
图1 普克鲁胺的结构式
3-氟-4-异硫氰酸根-2-三氟甲基苯甲腈(16)为合成Proxalutamide的重要中间体,而2-氟-3-三氟甲基苯胺(11)为合成Proxalutamide的关键前体化合物。目前,国内外关于11的合成工艺的研究报道较少,阻碍了Proxalutamide的大规模生产与应用。1989年,Wollweber[8]报道了以2-氟-5-氯-三氟甲苯为原料,经硝化得到2-氟-5-氯-3-硝基-三氟甲苯,再经催化氢化合成11。然而该工艺条件下的收率仅为50%,并且存在原料不易得、操作条件苛刻和危险系数较高等不利因素,因而不利于工业化生产。2021年,刘辉等[9]以邻氟三氟甲苯为原料,经叠氮化反应得到2-氟-3-三氟甲基苯基叠氮,再经雷尼镍、氢气催化加氢还原合成11。该工艺虽然合成步骤较少,但存在高温高压操作条件带来的潜在危险。
本文设计了一条合成16的新路线(图2)。以2-氟三氟甲苯为原料,经羧基化反应生成2-氟-3-三氟甲基苯甲酸(1a);1a经酰胺化反应生成2-氟-3-三氟甲基苯甲酰胺(1b);1b经Hofmann重排反应合成2-氟-3-三氟甲基苯胺(11)。11再经溴代合成12,12经氰基取代合成13,14,接着经水解合成15及氨基氧化合成最终产物Proxalutamide中间体3-氟-4-异硫氰酸根-2-三氟甲基苯甲腈(16),总收率36%。该合成路线的所有反应均为常压反应,操作简易安全,原料及试剂易于获取,同时各步反应收率和重复性较好。
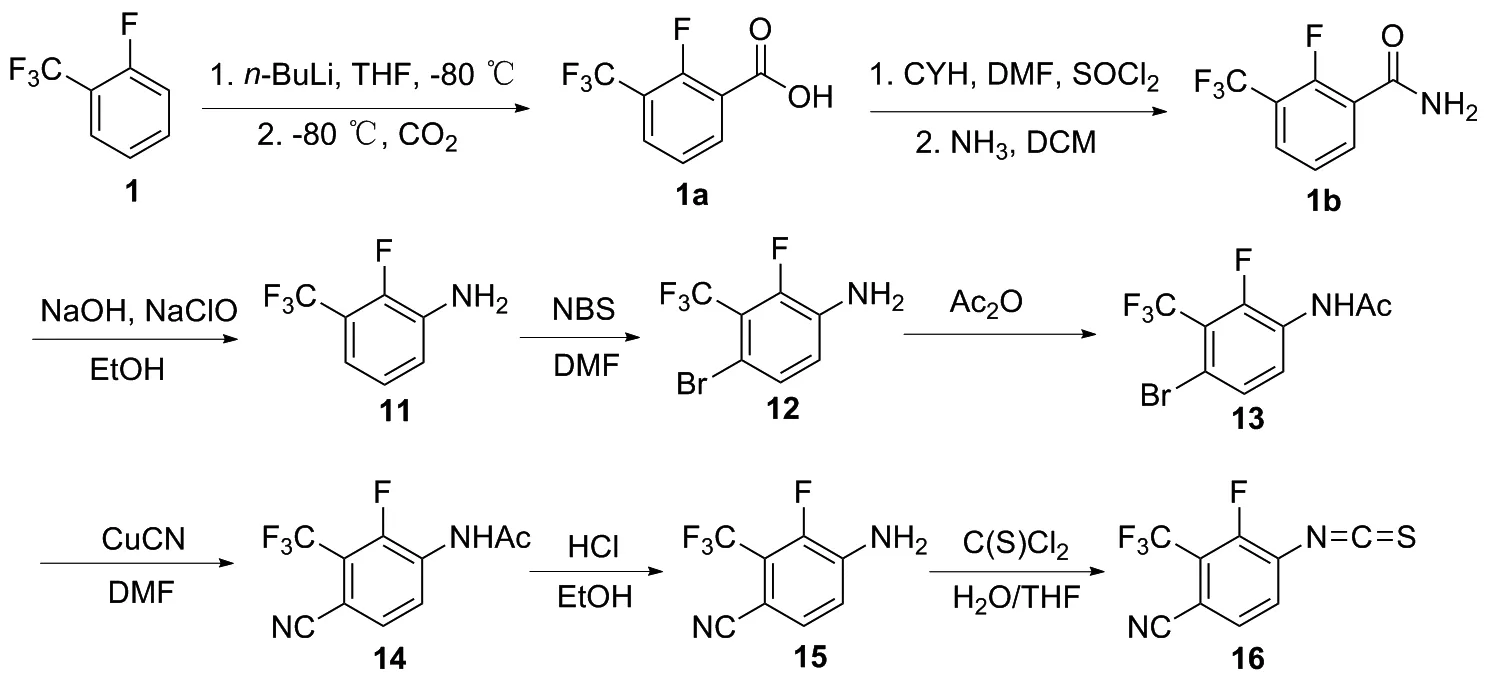
图2 化合物16的合成路线
1 实验部分
1.1 仪器与试剂
WRS-1B型数字熔点仪;Bruker Avance 500 MHz型核磁共振仪(TMS为内标,DMSO-d6或CDCl3为溶剂)。
所用试剂均为分析纯。
1.2 合成
(1) 2-氟-3-三氟甲基苯甲酸(1a)的合成
向250 mL圆底烧瓶中依次加入邻氟三氟甲苯(5.00 g, 30.49 mmol)和四氢呋喃(35 mL),氮气保护。温度降至-80 ℃,滴加浓度为2.70 mol/L正丁基锂的正己烷溶液(12.40 mL, 33.52 mmol),滴加完毕后保温反应5 h,并于-70 ℃条件下向反应体系中通入二氧化碳,鼓泡反应20 min。温度升高至-30 ℃,加入20 mL去离子水淬灭残余正丁基锂,温度升至室温,静置分层,分出水相,水相减压脱溶至无共沸,加入盐酸调节pH=2左右,有白色固体析出。于2~8 ℃条件下析晶0.5 h,抽滤,将滤饼于60 ℃条件下干燥,得白色固体化合物1a6.07 g,收率95.6%, m.p.126~128 ℃;1H NMR(500 MHz, DMSO-d6)δ: 13.74(s, 1H), 8.16(t,J=7.5 Hz, 1H), 7.99(t,J=7.0 Hz, 1H), 7.50(t,J=7.8 Hz, 1H);13C NMR(126 MHz, DMSO-d6)δ: 163.93, 159.22, 136.58, 131.29, 124.97, 123.55, 121.38, 121.17。
安徽省秸秆总量约4200万吨,其中水稻秸秆约1490万吨,小麦秸秆约1200万吨、玉米秸秆约400万吨,大豆秸秆约230万吨[12]。
(2) 2-氟-3-三氟甲基苯甲酰胺(1b)的合成
向100 mL烧瓶中加入1a(2.00 g, 9.61mmol),环己烷(10 mL), SOCl2(1.49 g, 12.49 mmol)和N,N-二甲基甲酰胺(DMF, 0.035 g, 0.48 mmol)),并于80 ℃条件下回流并保温反应5 h。冷却至室温,减压浓缩得到2-氟-3-三氟甲基苯甲酰氯粗品。向2-氟-3-三氟甲基苯甲酰氯粗品中加入二氯甲烷(DCM, 20 mL),冰水浴控制体系温度不超过15 ℃,通入氨气10 min。加入20 mL去离子水使体系中的盐溶解,搅拌静置分层,分出有机相。采用DCM(20 mL)萃取水相,并与有机相合并,真空浓缩得到白色固体化合物1b1.90 g,收率95.0%, m.p. 94~97 ℃;1H NMR(500 MHz, CDCl3)δ: 8.33(t,J=7.6 Hz, 1H), 7.79(t,J=7.2 Hz, 1H), 7.39(t,J=7.9 Hz, 1H), 6.63(br, 1H), 6.06(br, 1H);13C NMR(126 MHz, CDCl3)δ: 163.98, 159.15, 136.35, 130.96, 124.79, 123.38, 122.13, 121.22。
(3) 2-氟-3-三氟甲基苯胺(11)的合成
向100 mL圆底烧瓶中加入1b(2.00 g, 9.66 mmol),再加入30 mL乙醇搅拌溶解。加入质量分数为10%的NaOH溶液(4.25 g, 21 mmol),待体系温度降至0 ℃,开始滴加浓度为1.37 mol/L NaClO溶液(7.75 mL, 10.6 mmol),滴加完毕,保温反应40 min。升温至80 ℃,水解脱羧,回流反应5 h。冷却至室温,加入50 mL去离子水使盐溶解至出现乳化,采用乙酸乙酯(3×30 mL)萃取有机相并合并有机相,盐水(2×30 mL)洗涤、无水Na2SO4干燥、过滤,真空浓缩得到化合物111.61 g,收率92.3%;1H NMR(500 MHz, CDCl3)δ: 7.02~6.92(m, 3H), 3.89(br, 2H);13C NMR(126 MHz, CDCl3)δ: 149.42, 135.75, 124.28, 121.92, 120.46, 118.36, 115.50。
(4) 4-溴-2-氟-3-三氟甲基苯胺(12)的合成
在100 mL圆底烧瓶中将1(2.00 g, 11.17 mmol)溶于DMF(14 mL)中,并在15 ℃条件下分次加入N-溴代琥珀酰亚胺(NBS, 1.99 g, 11.17 mmol)),保温反应时间1 h。反应混合物用乙酸乙酯(EtOAc, 50 mL)稀释,并用水(2×50 mL)和饱和NaCl溶液(50 mL)洗涤。分离出的有机相用Na2SO4干燥、浓缩得到化合物粗品。柱层析纯化分离(洗脱剂:石油醚 ∶乙酸乙酯=10 ∶1,V∶V)得到化合物122.74 g,收率95.0%;1H NMR(500 MHz, DMSO-d6)δ: 7.31(d,J=8.7 Hz, 1H), 6.92(t,J=8.9 Hz, 1H), 5.81(s, 2H);13C NMR(126 MHz, DMSO-d6)δ: 149.41, 138.28, 131.00, 124.00, 121.82, 120.32, 102.02。
(5)N-[4-溴-2-氟-3-(三氟甲基)苯基]乙酰胺(13)的合成
向100 mL圆底烧瓶中加入化合物12(2.45 g, 9.5 mmol))和乙酸酐(6 mL)的混合物,并在25 ℃条件下搅拌反应3 h,减压蒸馏除去剩余乙酸酐。残留物加入冰水(10.00 g)和碳酸氢钠(pH=7)。用乙酸乙酯(3×30 mL)萃取混合物,合并有机相,Na2SO4干燥,过滤、浓缩得到化合物132.52 g,收率88.0%, m.p.102~103 ℃;1H NMR(500 MHz, DMSO-d6)δ: 10.06(s, 1H), 8.17(t,J=8.4 Hz, 1H), 7.68(d,J=8.9 Hz, 1H), 2.12(s, 3H);13C NMR(126 MHz, DMSO-d6)δ: 169.21, 152.16, 130.71, 127.81, 127.71, 123.15, 120.98, 112.72, 23.57。
(6)N-[4-氰基-2-氟-3-(三氟甲基)苯基]乙酰胺(14)的合成
(7) 4-氨基-3-氟-2-(三氟甲基)苯甲腈(15)的合成
向100 mL圆底烧瓶中依次加入化合物14(0.70 g, 2.84 mmol)的乙醇(5 mL)溶液和浓盐酸(12 mol/L, 5 mL)。将混合物溶液在78 ℃条件下回流反应1 h,冷却至室温,真空浓缩,所得固体溶于乙酸乙酯(25 mL),用饱和碳酸氢钠溶液(25 mL)洗涤,无水MgSO4干燥,过滤、浓缩后得到淡灰色固体化合物150.54 g,收率93.0%, m.p.160~162 ℃;1H NMR(500 MHz, DMSO-d6)δ: 7.55(d,J=8.5 Hz, 1H), 7.04(t,J=8.5 Hz, 1H), 6.83(s, 2H);13C NMR(126 MHz, DMSO-d6)δ: 147.56, 142.91, 132.29, 123.01, 120.83, 118.31, 116.75, 91.94。
(8) 3-氟-4-异硫氰酸根-2-三氟甲基苯甲腈(16)的合成
在100 mL圆底烧瓶中将化合物15(0.41 g, 2 mmol)溶于四氢呋喃(THF, 5 mL),并在20 ℃条件下缓慢加入硫光气(2 mL)水(5 mL)溶液。混合物搅拌反应1 h后浓缩。残留物在水(50 mL)和乙酸乙酯(30 mL)之间分配。水相用乙酸乙酯(2×30 mL)萃取,合并有机相,用盐水(2×50 mL)洗涤,无水MgSO4干燥,过滤、浓缩,得到黄色油状液体化合物160.40 g,收率73.0%;1H NMR(500 MHz, CDCl3)δ: 7.61(d,J=8.3 Hz, 1H), 7.44(t,J=7.7 Hz, 1H);13C NMR(126 MHz, CDCl3)δ: 157.60, 146.58, 131.41, 129.50, 127.97, 121.97, 119.77, 114.40, 108.62。
2 结果与讨论
2.1 合成1a的反应条件优化
合成1a的反应为邻氟三氟甲苯的邻位金属化反应,通过相关文献[10-13]发现,正丁基锂用量、溶剂用量及反应时间对收率均存在一定影响。为了获得较佳的反应条件,本文考察了正丁基锂用量和反应时间对反应收率的影响。
(1) 正丁基锂用量对1a收率的影响
以5.00 g1为原料,50 mL THF为溶剂,设置反应时间为5 h,分析正丁基锂用量对1a收率的影响,实验结果如表1所示。由表1可知,随着正丁基锂用量的加大,1a收率不断增长。当正丁基锂的用量为1.1 eq.时,产物1a可以获得较佳收率(92.0%),再继续增加正丁基锂的用量,收率趋于稳定,变化不明显。

表1 正丁基锂用量对1a收率的影响
(2) 反应时间对1a收率的影响
以5.00 g1为原料,正丁基锂用量为1.1 eq., THF为50 mL,分析反应时间对1a收率的影响,实验结果如表2所示。由表2可知,随着反应时间的增长,收率增大。当反应时间为5 h(95.6%)时,收率趋于稳定。

表2 反应时间对1a收率的影响
2.2 合成1b的反应条件优化
合成1b的反应为羧酸的酰氯、酰胺化反应,酰氯化指将含酰基的物质(羧基或酸酐)与含活泼氯的试剂反应,生成酰氯的过程[14]。酰氯是羧酸衍生物中最活泼的酰基化试剂,酰氯氨解可制备酰胺。据文献[15-17]报道,影响反应收率的因素有酰氯化试剂、反应溶剂、催化剂种类和反应温度等。本文主要考察酰氯化试剂种类和反应溶剂体系对收率的影响。
(1) 酰氯化试剂种类对1b收率的影响
以2.00 g1a为原料,酰氯化试剂均为理论量的2倍量,催化剂DMF用量为0.05 eq.,溶剂二氯甲烷为20 mL,分析不同酰氯化试剂对1b收率的影响,实验结果如表3所示。由表3可知,当选择氯化亚砜为酰氯化试剂时,产物1b收率最高。虽草酰氯氯化能力更强,能获得与氯化亚砜相当的产物收率,但草酰氯毒性更大,价格昂贵,因此本文选择氯化亚砜作为酰氯化试剂。

表3 酰氯化试剂种类对1b收率的影响
(2) 反应溶剂对1b收率的影响
以2.00 g1a为原料,氯化亚砜用量为2.0 eq.,催化剂DMF用量为0.05 eq.,氯化亚砜与溶剂的质量体积比(m/V)为1 ∶10,考察不同溶剂体系对1b收率的影响,实验结果如表4所示。由表4可知,当溶剂为环己烷时,产物1b收率最佳(94.0%)。

表4 反应溶剂对1b收率的影响
2.3 合成11的反应条件优化
合成11的反应为Hofmann重排反应,于1881年由Hofmann[18]提出。经典反应条件以一级酰胺为起始物,在次卤酸盐和碱的作用下,经异氰酸酯中间体得到比起始物少1个碳原子的伯胺,所以又称霍夫曼降解反应。霍夫曼重排反应过程中主要存在以下几种副反应:原料酰胺的水解、产物的过度卤化和脲及酰基脲的生成等[19]。本文考察了NaClO用量和NaOH用量对反应收率的影响。
(1) NaClO溶液用量对11收率的影响
以2.00 g1b为原料,30 mL乙醇为溶剂,NaOH用量为2.2 eq,重排反应温度为0 ℃,重排反应时间为40 min,水解脱羧温度为80 ℃,水解脱羧时间为5 h,分析NaClO溶液用量对11收率的影响,实验结果如表5所示。由表5可知,当NaClO溶液用量为1.1 eq时,11收率最高(92.3%),而继续增大NaClO溶液用量,11收率下降,该现象可能是NaClO溶液用量过多,导致产物的过度卤化,副产物含量增大。

表5 NaClO溶液用量对11收率的影响
(2) NaOH用量对11收率的影响
以2.00 g1b为原料,30 mL乙醇为溶剂,NaClO用量为1.1 eq,重排反应温度为0 ℃,重排时间为40 min,水解脱羧温度为80 ℃,水解脱羧时间为5 h,考察NaOH用量对11收率的影响,实验结果如表6所示。由表6可知,随着NaOH用量的增大,产率先增大而后减小。当NaOH用量为2.2 eq时,11收率最高,这是由于异氰酸酯非常活泼,当增加NaOH用量时,生成的中间体异氰酸酯可以很快转化为伯胺,但如果NaOH用量过大,N-氯代酰胺可以发生盐析作用,导致胺化程度开始呈下降趋势。

表6 NaOH用量对11收率的影响
本文以邻氟三氟甲苯为原料合成关键前体化合物11,再经溴代、氰基取代、水解和氧化得到Proxalutamide中间体3-氟-4-异硫氰酸根-2-三氟甲基苯甲腈,并对11的合成条件进行了优化,总收率为36%。与已有合成路线相比,该合成路线的原料及试剂易得,所有反应均为常压反应,反应条件温和且操作简易安全,具有较好的工业化前景。
猜你喜欢
山东化工(2024年1期)2024-02-04 09:47:12
能源化工(2021年2期)2021-12-30 18:31:06
分析化学(2019年3期)2019-03-30 10:59:24
广东石油化工学院学报(2016年6期)2016-05-17 05:17:25
生物化工(2016年4期)2016-04-08 10:26:27
合成化学(2015年10期)2016-01-17 08:56:30
山东工业技术(2015年6期)2015-07-27 00:53:22
合成化学(2015年5期)2015-03-26 06:02:22
有机氟工业(2014年3期)2014-06-05 14:36:38
石油化工应用(2014年2期)2014-03-11 17:38:59
