
螺[3,2′]吡咯烷氧化吲哚的化学拆分
2023-08-19 00:30黄志诚宋祥家王立新
合成化学 2023年8期
黄志诚, 邹 滢, 宋祥家, 田 芳, 王立新
(1. 中国科学院 成都有机化学研究所,四川 成都 610041; 2. 中国科学院大学,北京 100049)
螺[3,2′]吡咯烷氧化吲哚是众多生理活性物质及天然产物的核心骨架[1-3]。近年来,药物化学家们发现了大批具有抗菌、抗炎、抗病毒以及抑制癌细胞生长生理活性的螺[3,2′]吡咯烷螺环氧化吲哚类化合物[4-6]。该类化合物的主要构建方案包括基于3-氨基氧化吲哚偶极子环的加成反应[7-8]以及基于氧化吲哚底物的串联反应[9-10]。这两类反应构建的螺[3,2′]吡咯烷螺环氧化吲哚化合物的吡咯烷结构单元会引入单个或多个吸电子基团,从而诱导加成环化反应及串联反应。而吡咯烷结构单元不存在取代基螺[3,2′]吡咯烷氧化吲哚,难以用上述方法进行构建。
基于对手性螺环氧化吲哚的前期研究[11-13],2019年,本课题组继续发展了一种高效的消旋体合成方法[11](图1)。对消旋体的化学拆分是获得手性化合物最广泛的方案[14-15],因而本文采用二对甲苯酰基-L-酒石酸[(-)-DDTA]为拆分剂,以43%收率以及94%ee成功实现了1-苄基螺[3,2′]吡咯烷氧化吲哚的化学拆分(图2)。产物结构经1H NMR和13C NMR表征。
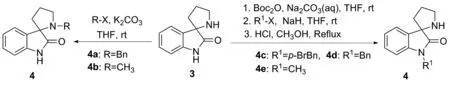
图1 螺[3,2′]吡咯烷氧化吲哚的衍生策略

图2 螺[3,2′]吡咯烷氧化吲哚的化学拆分
1 实验部分
1.1 仪器与试剂
Brucker-300型核磁共振仪;日本岛津LC-20AT型高效液相色谱仪。
所用化学试剂均为分析纯。
1.2化合物4a~4b的合成(以4a为例)
在装有20.00 mL THF的50.00 mL单口烧瓶中依次加入1.88 g(1.00 mmol)螺环氧化吲哚3、 1.66 g(12.00 mmol) K2CO3以及等当量的氯化苄。室温下搅拌至化合物3反应完全(TLC检测)。旋干反应溶剂并加入15.00 mL蒸馏水,乙酸乙酯(3×10.00 mL)萃取水相,合并有机相,无水Na2SO4干燥。采用柱层析(洗脱剂:EA ∶PE=2 ∶1,V∶V)分离纯化得到目标产物4a。采用类似的方法合成化合物4b。
1′-苄基螺[3,2′]吡咯烷氧化吲哚4a:白色固体1.75 g,收率78%, m.p.105~107 ℃;1H NMRδ: 10.30(s, 1H), 7.33(dd,J1=7.4 Hz,J2=1.3 Hz, 1H), 7.28~7.12(m, 6H), 7.00(td,J1=7.5 Hz,J2=1.1 Hz, 1H), 6.87~6.75(m, 1H), 3.41(s, 1H), 3.28(s, 1H), 2.94(m, 2H), 2.17~1.92(m, 4H);13C NMRδ: 179.8, 142.5, 139.3, 130.8, 128.7, 128.1, 128.0, 126.8, 123.8, 122.0, 109.5, 70.9, 53.1, 50.4, 35.8, 21.7; HR-MS(ESI)m/z: calcd for C18H19N2O+{[M+H]+}279.1492, found, 279.1495。
1′-甲基螺[3,2′]吡咯烷氧化吲哚4b:白色固体1.58 g,收率87%, m.p.89~91 ℃;1H NMRδ: 9.21(s, 1H), 7.29~7.15(m, 2H), 7.02(td,J1=7.5 Hz,J2=1.1 Hz, 1H), 6.88(dt,J1=7.8 Hz,J2=0.8 Hz, 1H), 3.41~3.23(m, 1H), 3.15(m, 1H), 2.36~2.08(m, 7H);13C NMRδ: 181.5, 141.2, 130.6, 128.6, 124.2, 122.6, 110.0, 71.8, 54.0, 36.4, 35.5, 22.3; HR-MS(ESI)m/z: calcd for C18H19N2O+{[M+H]+}203.1179, found, 203.1173。
1.3 化合物4c~4e的合成(以4d为例)
向圆底烧瓶中加入9.40 g(5.00 mmol)螺环氧化吲哚3,然后依次加入13.10 g(6.00 mmol)二碳酸二叔丁酯(Boc2O), 40.00 mL 四氢呋喃(THF)以及20.00 mL饱和Na2CO3溶液。常温下剧烈搅拌约6 h至反应完全(TLC检测)。加入30.00 mL氯化铵溶液淬灭反应,再用乙酸乙酯(3×15.00 mL)萃取,合并有机相,用20.00 mL饱和食盐水洗涤,经无水硫酸钠干燥后浓缩得到N-Boc螺环氧化吲哚。将2.88g(1.00 mmol)N-Boc吡咯烷氧化吲哚溶于20.00 mL THF,并在冰水浴下分批加入0.45 g(1.10 mmol) 60% NaH矿物油混合物。随后加入1.39 g(0.81 mmol)溴化苄,20 h后反应完全(TLC检测)。加入氯化铵溶液淬灭反应,再加入20.00 mL蒸馏水,二氯甲烷萃取,浓缩得到淡黄色固体。甲醇结晶得到2.82 g(0.75 mmol)白色固体。随后将得到的固体置于圆底烧瓶中,加入20.00 mL甲醇盐酸溶液回流,期间有大量白色固体生成,2 h反应完全(TLC检测)。减压蒸馏除去溶剂并加入20.00 mL水,白色固体溶解,二氯甲烷(2×10.00 mL)洗涤水相,随后利用1.00 M氢氧化钠溶液调节水相pH=12~13,期间有大量固体生成,乙酸乙酯(3×20.00 mL)萃取,浓缩后以93%收率得到1.42 g(0.51 mmol)4d。通过与CDCC: 2075368及其对应的旋光值进行对比,最终确定了化合物4d的绝对构型为S。采用类似的方法合成化合物4c和4e。
1-对溴苄基螺[3,2′]吡咯烷氧化吲哚4c:无色油状液体1.88 g,收率86%;1H NMRδ: 7.43~7.31(m, 2H), 7.25(dd,J1=7.5 Hz,J2=1.3 Hz, 1H), 7.15~7.03(m, 3H), 6.95(td,J1=7.5 Hz,J2=1.1 Hz, 1H), 6.60(d,J=7.7 Hz, 1H), 4.85~4.62(m, 2H), 3.55~3.08(m, 2H), 2.33~1.76(m, 5H);13C NMRδ: 179.7, 174.2, 141.4, 134.5, 133.2, 131.5, 131.4, 128.6, 128.5, 128.2, 122.8, 122.7, 121.1, 108.6, 67.0, 47.5, 42.7, 37.5, 25.8, 21.2; HR-MS(ESI)m/z: calcd for C18H18N2OBrNa+{[M+Na]+}Br79: 379.0417, Br81: 381.0396, found, Br79: 379.0414, Br81: 381.0400。

1-甲基螺[3,2′]吡咯烷氧化吲哚4e:无色油状液体1.63 g,收率90%;1H NMRδ: 7.26(m,J1=7.7 Hz,J2=6.6 Hz,J3=1.5 Hz, 2H), 7.05(td,J1=7.5 Hz,J2=1.0 Hz, 1H), 6.88~6.73(m, 1H), 3.51~3.38(m, 1H), 3.37~3.24(m, 1H), 3.18(s, 3H), 2.25~1.94(m, 5H);13C NMRδ: 180.4, 143.0, 134.5, 128.5, 122.9, 122.7, 108.0, 67.5, 48.1, 37.6, 26.3, 26.2; HR-MS(ESI)m/z: calcd for C12H15N2O+{[M+H]+}203.1179, found, 203.1178。
1.4 化合物4d的拆分
将合成的酰胺N-Bn螺环吡咯烷氧化吲哚4d1.42 g(0.51mmol)溶于10.00 mL无水乙醇,之后加入2.12 g(0.55 mmol) (-)-DDTA5b,于75 ℃条件下回流约30 min,有大量白色固体析出。分次逐渐加入无水乙醇5.00 mL,体系固体完全溶解。停止加热,冷却至室温,抽滤分离固体。用1.00 M氢氧化钠溶液游离仲胺盐,乙酸乙酯(3×10.00 mL)萃取,合并有机相,无水硫酸钠干燥,旋干得0.61 gN-Bn吡咯烷螺环氧化吲哚ee94%。
无取代基的手性螺环胺与多种手性拆分试剂成盐结晶都不存在拆分效果,因此,本研究对化合物进行衍生。首先利用仲胺的亲核性,构建1′-苄基取代的螺环叔胺4a以及1′-甲基取代的叔胺4b。研究结果表明,两种化合物均无法与手性拆分试剂成盐,这可能是由于叔胺氮原子靠近螺环手性中心,螺环手性中心的刚性太强,空间位阻较大,从而阻碍了手性拆分试剂与仲胺的结合。之后本课题组对氧化吲哚酰胺氮原子上的取代基进行衍生,分别合成了1-对溴苄基螺环胺4c、 1-苄基螺环胺4d以及1-甲基螺环胺4e。在化合物拆分过程中发现,化合物4c与手性酸成盐后不能生成固体,之后对化合物4d和4e的拆分条件进行优化,结果如表1所示。由表1中Entries 1~3可知,以一当量的化合物5b为拆分试剂,甲醇为析晶溶剂时,化合物4d拆分效果较好。通过对溶剂进行筛选,最终确定在(-)-DDTA为拆分试剂的条件下以43%收率以及94%ee成功实现化合物4d的化学拆分(Entry 4)。然而,通过对化合物4e的拆分条件进行研究后发现,化合物4e在各种拆分试剂的作用下都未存在明显的拆分效果(Entries 6~10)。
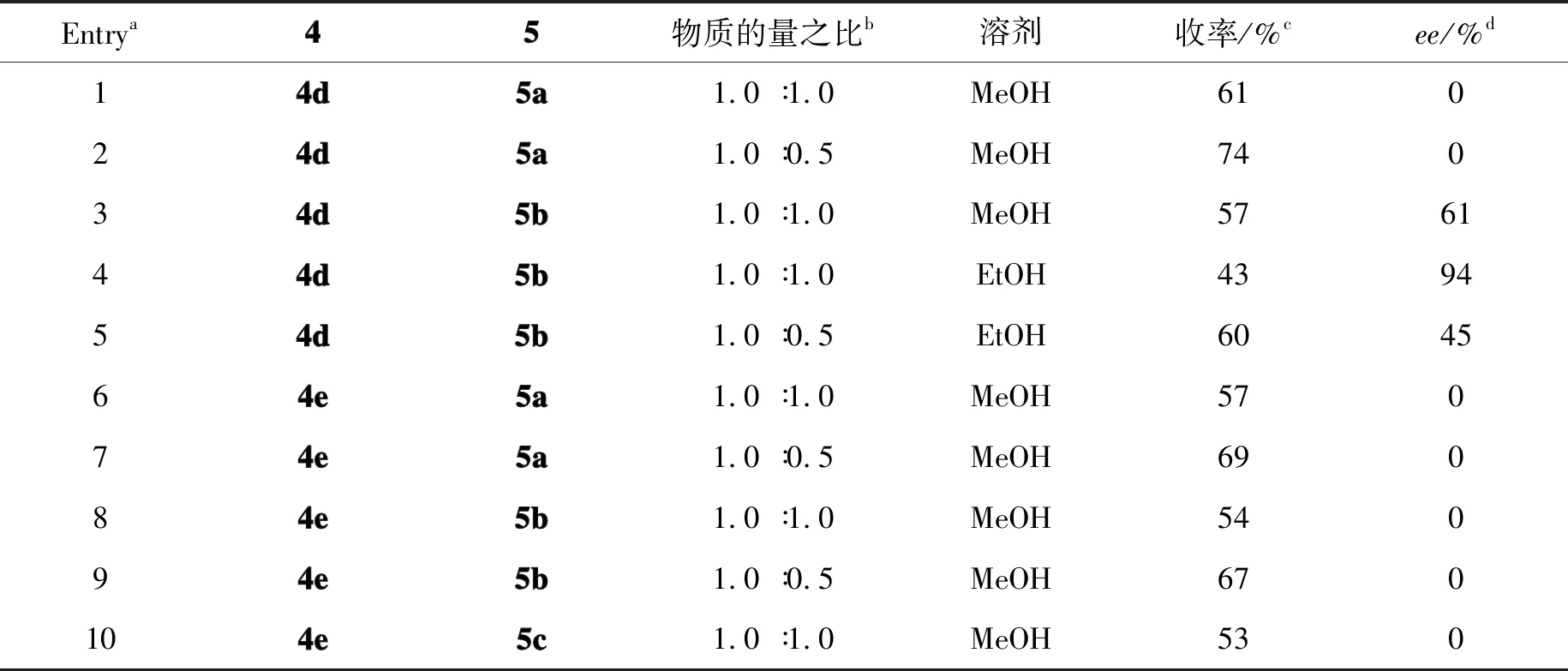
表1 螺[3,2′]吡咯烷氧化吲哚的化学拆分
本文对螺[3,2′]吡咯烷氧化吲哚的化学拆分进行了研究,确定了1-苄基螺[3,2′]吡咯烷氧化吲的最佳拆分条件为:以(-)-DDTA为拆分试剂,在乙醇中重结晶。该研究最终以43%收率,94%ee成功实现化学拆分。通过与已知手性单晶结构的对比确定了拆分产物的绝对构型为S。
猜你喜欢
分子催化(2022年1期)2022-11-02
化工时刊(2020年4期)2020-06-07
化工管理(2017年26期)2017-03-04
国外医药(抗生素分册)(2016年4期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
合成化学(2016年5期)2016-06-13
合成化学(2015年10期)2016-01-17
合成化学(2015年1期)2016-01-17
分析测试学报(2015年5期)2016-01-13
中国洗涤用品工业(2015年9期)2015-02-28
