
一种靶向DAT的氘代正电子探针的制备与表征
2023-08-19 00:30胡潜岳刘春仪陈正平
合成化学 2023年8期
胡潜岳, 唐 婕, 方 毅, 刘春仪, 陈正平,*
(1. 南京医科大学 药学院,江苏 南京 211166; 2. 江苏省原子医学研究 国家卫生健康委员会核医学重点实验室,江苏省分子核医学重点实验室,江苏 无锡 214063)
中枢神经系统的多巴胺转运体(DAT)是许多神经退行性疾病的潜在靶点,如帕金森病(PD)[1]、精神分裂症[2]和抑郁症[3]等。脑内DAT数量的变化可以间接反映脑内多巴胺能神经元的变化。为了使DAT可视化,研究者已经开发了各种成像技术,如正电子发射断层扫描(PET)是一种无创的成像技术,具有灵敏度高,特异性强及安全性好的优点。而研发正电子核素(如11C、18F)标记的靶向DAT的分子探针进行PET成像有助于对相关疾病的诊断、疗效监测和疾病机理研究。
目前,大量11C或18F标记的放射性探针已被开发用于PET成像[4]。与11C标记的探针相比,18F标记的探针具有更低的正电子能量和合适的半衰期,是广泛用于PET成像的正电子核素。靶向DAT的探针[18F]FP-CIT是目前国内外临床应用最广泛的18F标记的正电子探针,因具有半衰期适宜、亲和力和特异性高等优点,被用于PD和多种与多巴胺能神经变性相关的成像,在临床中应用广泛[5]。然而,[18F]FP-CIT在体内细胞色素P450酶的作用下,N-氟丙基易代谢脱烷基而产生[18F]氟烷基代谢物,这些放射性代谢产物能够进脑,从而影响PET成像的准确性[6]。因此,开发出体内稳定性高、DAT靶向性好的PET探针,将有助于PET技术对脑内DAT进行精准成像和定量分析,从而更有利于对DAT相关疾病的基础与临床研究。
将药物易代谢物部位的C—H用C—D取代从而制得氘代药物,能够降低药物在体内的代谢速度,提高体内稳定性,改善药代动力学。氘代技术已在某些药物(例如Austedo)取得了成功的应用[7]。在放射性药物领域,已有文献报道了几个成功的氘代探针[8-10]。基于此,本研究利用氘代策略,设计并合成了一种[18F]FP-CIT的氘代结构[18F]FP-CIT-d6([18F]7),预期新氘代探针[18F]7在保持DAT靶向性的同时,还能减缓探针的体内代谢速度,减少放射性代谢产物的产生,从而提高PET显像的准确性。化学合成方法为:利用丙二酸二乙酯(化合物1)在重水中通过氘氢交换得到化合物2,用氘代氢化铝锂还原化合物2得到氘代丙二醇(化合物3),化合物3再经过磺酰化制得磺酸酯(化合物4),化合物4和四丁基氟化铵(TBAF)发生亲核取代反应得到化合物5,化合物5与化合物6反应得到非放射性靶向DAT的化合物FP-CIT-d6(7);化合物3经磺酰化反应后得到化合物8,化合物8与化合物6进行亲核取代反应得到化合物9,再与甲基磺酸酐发生磺酰化反应得到标记前体化合物10。采用质谱、核磁共振氢谱、碳谱和氟谱对合成的中间体、非放射性化合物与标记前体化合物进行了结构表征。利用前体化合物10与18F-在碱性环境中进行一步法标记得到放射性探针[18F]FP-CIT-d6([18F]7);利用放射性HPLC测定了其放射性化学纯度,并验证放射性探针的结构。本文还测定了探针[18F]7在磷酸盐缓冲液(PBS)和胎牛血清(FBS)中的体外稳定性,用摇瓶法测定了[18F]7脂水分配系数(LogP),为探针后续体内生物活性研究提供基础。
1 实验部分
1.1 仪器与试剂
Avance III 400MHz型核磁共振仪(溶剂:氘代氯仿,CDCl3;内标物:四甲基硅烷,TMS);SQ Detector 2型电子喷雾离子质谱仪;住友H7型回旋加速器;Waters 1525 Binary型高效液相色谱仪;Waters 1525 Plus Binary型半制备型高效液相色谱仪;Waters 2998型双通道紫外可见光检测器;Gabi Nova型放射检测器。
丙二酸二乙酯(化合物1)、重水(D2O)、氘代氢化铝锂(LiAlD4)、对甲苯磺酰氯(TsCl)、 4-二甲氨基吡啶(DMAP)、吡啶(Py)、四丁基氟化铵(TBAF)、甲基磺酸酐(Ms2O)、 2-甲基-2-丁醇(TAA)均购自安耐吉化学试剂有限公司;乙醚(分析纯)、正己烷(分析纯)、乙酸乙酯(分析纯)、四氢呋喃(THF)、甲苯(PhMe,分析纯)、二氯甲烷(CH2Cl2,分析纯)均购自国药集团化学试剂有限公司;乙腈(CH3CN,色谱纯)、甲醇(色谱纯)、三乙胺(TEA,色谱纯)、三氟乙酸(TFA,色谱纯)均购自北京百灵威科技有限公司;2β-甲酯基-3β-(4-碘苯基)去甲基托烷(化合物6)参照文献[11-12]自制。
1.2 合成
非放射性化合物7和标记前体化合物10的合成路线如图1~2所示。
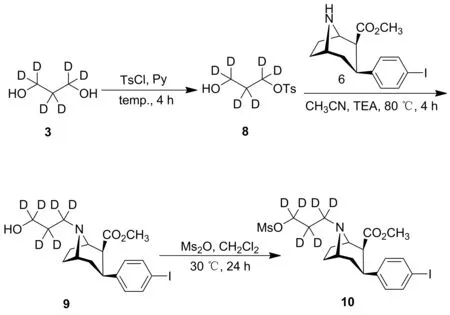
图2 标记前体化合物10的合成路线
(1) 化合物2的合成
化合物2参照文献[13]步骤合成,在丙二酸二乙酯(化合物1, 1.60 g, 10.00 mmol)的D2O(20.0 mL)加入无水碳酸钾(K2CO3, 0.14 g, 1.00 mmol),反应液在室温下搅拌24 h,反应结束后用乙醚萃取,有机相用无水硫酸钠干燥,除去溶剂后得到1.26 g化合物2:无色透明液体,产率78%;1H NMR(400 MHz, CDCl3)δ: 4.13(q,J=7.2 Hz, 4H), 1.21(t,J=7.2 Hz, 6H);13C NMR(101 MHz, CDCl3)δ: 166.48, 61.30, 41.06(quint.,JC,D=20.1 Hz), 13.92; ESI-MSm/z: Calcd for C7H10D2NaO4{[M+Na]+} 185.08, found 185.24。
(2) 化合物3的合成
化合物2(1.94 g, 11.97 mmol)的3 mol/L THF溶液在0 ℃下,缓慢滴加到1.0 mol/L LiAlD4(0.60 g, 14.36 mmol)的THF溶液中,反应液加热回流8 h,室温下搅拌过夜。缓慢滴加1.2 mol/L氢氧化钠的D2O(0.20 mmol)溶液,过滤沉淀,乙醚洗涤,滤液除去溶剂后得到0.49 g淡黄色油状液体化合物3,产率47%。
(3) 化合物4的合成
根据文献[14]报道的方法合成化合物4。将化合物3(0.67 g, 8.22 mmol)、 DMAP(0.10 g, 0.82 mmol)和TEA(3.30 g, 32.80 mmol)溶于CH2Cl2(10.0 mL)中,在0 ℃下滴加TsCl(3.84 g,20.22 mmol)的CH2Cl2(30.0 mL)溶液,反应液升至室温搅拌24 h,加饱和NaCl溶液(20.0 mL)淬灭反应,CH2Cl2(2×20.0 mL)萃取,有机相用无水硫酸钠干燥,过滤除去溶剂后得到白色固体,在90 ℃下用乙醇重结晶,得到化合物4:白色固体1.34 g,产率42%;1H NMR(600 MHz, CDCl3)δ: 7.75(d,J=8.3 Hz, 4H), 7.35(d,J=8.0 Hz, 4H), 2.46(s, 6H);13C NMR(151 MHz, CDCl3)δ: 145.09, 132.62, 129.98, 127.88, 21.66; ESI-MSm/z: Calcd for C17H14D6O6NaS2{[M+Na]+} 413.10, found 413.38。
(4) 化合物5的合成
将化合物4(0.67 g, 1.70 mmol)溶于CH3CN(8.0 mL)中,滴加TBAF(3.0 mL),加热至90 ℃反应2 h,除去溶剂后,利用硅胶柱层析(正己烷 ∶乙酸乙酯=4 ∶1,V∶V)分离纯化得到0.20 g化合物5:无色透明液体,产率50%,1H NMR(600 MHz, CDCl3)δ: 7.82~7.77(m, 2H), 7.36(d,J=7.6 Hz, 2H), 2.45(s, 3H);13C NMR(151 MHz, CDCl3)δ: 144.99, 132.86(d,JC,F=3.02 Hz), 129.95, 127.89, 78.8(m,JC,D=24.16 Hz), 65.51(m,JC,D=6.04 Hz), 29.05(quint,JC,D=19.63 Hz), 21.64;19F NMR(376 MHz, CDCl3)δ: -223.29; GC-MSm/z: Calcd for C7H7D6FO3S 238.09, found 238.1。
(5) 化合物7的合成
将化合物5(0.24 g, 1.01 mmol)、化合物6(0.31g, 0.82 mmol)、 TEA(0.8 mL)溶于30.0 mL甲苯中,在115 ℃下反应4 h,除去溶剂后通过柱层析(乙醚 ∶正己烷 ∶三乙胺=10.0 ∶7.0 ∶0.1,V∶V∶V)分离纯化得到0.19 g化合物7:白色固体,产率53%;1H NMR(400 MHz, CDCl3)δ: 7.57(d,J=8.1 Hz, 2H), 7.00(d,J=8.1 Hz, 2H), 3.66(d,J=3.9 Hz, 1H), 3.49(s, 4H), 3.01~2.73(m, 2H), 2.52(t,J=10.9 Hz, 1H), 2.15~1.91(m, 2H), 1.78~1.57(m, 3H);13C NMR(101 MHz, CDCl3)δ: 171.76, 143.03, 136.88, 129.49, 91.02, 63.12, 61.32, 52.67, 50.97, 33.89, 33.86, 26.03, 25.93;19F NMR(376 MHz, CDCl3)δ: -222.84; ESI-MSm/z: Calcd for C18H18D6FINO2{[M+H]+} 438.12, found 438.39。
(6) 化合物8的合成
根据文献[14]报道的方法合成化合物8。在0 ℃下,向化合物3(0.50 g, 6.00 mmol)的CH2Cl2(3.0 mL)溶液滴加TsCl(1.26 g, 6.60 mmol)和吡啶(0.5 mL, 6.60 mmol)的CH2Cl2(3.0 mL)溶液,反应液升至室温搅拌4 h,加饱和NaCl溶液(30.0 mL)淬灭反应,CH2Cl2(3×30.0 mL)萃取,有机相用无水硫酸钠干燥,过滤除去溶剂后通过硅胶柱层析(正己烷 ∶乙酸乙酯=50 ∶50,V∶V)纯化,得到0.75 g化合物8:无色透明油状液体,产率53%;1H NMR(400 MHz, CDCl3)δ: 7.75(d,J=8.0 Hz, 2H), 7.33(d,J=8.0 Hz, 2H), 2.61(dd,J=7.9 Hz, 4.4 Hz, 1H), 2.41(s, 3H);13C NMR(101 MHz, CDCl3)δ: 144.98, 132.78, 129.95, 127.80, 67.03(quint.,JC,D=23.2 Hz), 57.28(quint.,JC,D=22.1 Hz), 32.52(quint.,JC,D=19.5 Hz), 21.60; ESI-MSm/z: Calcd for C10H9D6O4S {[M+H]+}237.11, found 237.06。
(7) 化合物9的合成
将化合物6(0.07 g, 0.20 mmol)、化合物8(0.04 g, 0.15 mmol)、 TEA(15 μL)溶于CH3CN(3.0 mL)中,于80 ℃加热回流4 h,除去溶剂后,加CH2Cl2(5.0 mL)溶解残余物,再用5%氢氧化钠(5.0 mL)洗涤,收集有机相,除去溶剂后干燥得到0.05 g化合物9:淡黄色油状液体,产率69%;1H NMR(400 MHz, CDCl3)δ: 7.55(d,J=8.3 Hz, 2H), 6.96(d,J=6.5 Hz, 2H), 4.73(s, 1H), 3.61(d,J=17.8 Hz, 2H), 3.49(dd,J=3.4 Hz, 1.6 Hz, 3H), 2.97(dd,J=12.9, 5.0 Hz, 1H), 2.89(s, 1H), 2.51(t,J=12.7 Hz, 1H), 2.17~1.98(m, 2H), 1.78~1.57(m, 3H);13C NMR(101 MHz, CDCl3)δ: 172.08, 142.28, 136.98, 129.46, 91.31, 64.65, 59.32, 52.98, 51.39, 33.87, 33.61, 26.36, 24.91; ESI-MSm/z: Calcd for C18H19D6INO3{[M+H]+} 436.13, found 436.43。
(8) 化合物10的合成
将化合物9(0.05 mg, 0.10 mmol)溶解在CH2Cl2(0.5 mL)中,加入甲基磺酸酐(0.04 g, 0.23 mmol)。搅拌24 h后,用高效液相色谱监测。反应完全后加入2.0 mL乙醚沉淀,除去分离出的悬浊液,残余物用1.0 mL的CH2Cl2溶解,再次加入乙醚,重复洗涤3次。将残余物真空干燥,得到0.04 g甲磺酸盐前体化合物10:白色固体,产率72%;1H NMR(400 MHz, CDCl3)δ: 8.77(s, 1H), 7.64(d,J=7.9 Hz, 2H), 6.96(d,J=8.0 Hz, 2H), 4.32(s, 2H), 3.51~3.36(m, 4H), 3.17(s, 3H), 3.08(d,J=6.3 Hz, 1H), 2.86(s, 4H), 2.59(s, 1H), 2.44(s, 1H), 2.28~2.07(m, 2H), 1.96(d,J=14.7 Hz, 1H);13C NMR(101 MHz, Chloroform-d)δ: 173.99, 137.84, 137.78, 129.45, 93.29, 63.60, 62.24, 52.68, 49.06, 39.39, 37.24, 34.16, 31.84, 24.47, 23.56; ESI-MSm/z: Calcd for C19H20D6INO5S {[M+H]+} 514.10, found 514.29。标记前体化合物10的高效液相色谱和质谱如图3所示。根据图4中标记前体化合物10的核磁共振氢谱所知,δ值为8.77(s,1H), 3.17(s, 3H)处的信号峰为甲磺酸盐上的氢;图5中核磁共振碳谱δ值为39.39处的信号峰对应甲磺酸盐上的碳。

m/z

δ

δ
1.3 探针[18F]7的放射性合成
探针[18F]7的放射性合成路线如图6所示。回旋加速器产生18F离子经氮气传输被吸附在阴离子交换柱(QMA)上,用1.1 mL淋洗液(含15 mg K222和3 mg K2CO3的10%乙腈水溶液)洗脱18F离子至反应管中,乙腈干燥2次后,加入4 mg标记前体化合物10(溶解于100 μL CH3CN和1.0 mL TAA中),100 ℃反应20 min。反应结束后采用半制备型HPLC分离,液相条件为甲醇∶水∶三乙胺=75.0 ∶25.0 ∶0.1(V∶V∶V),接峰组分用水稀释,通过C18柱吸附标记探针[18F]7,洗去多余的18F离子,再用2.0 mL乙醇淋洗进西林瓶,即得纯化后探针[18F]7的乙醇溶液。取探针[18F]7(10 μCi, 5 μL)与化合物7(5 μL)共进样,用配备放射性检测器和紫外检测器的HPLC分析其结构和放射性化学纯度。

图6 探针[18F]7的放射合成路线
1.4 探针[18F]7的体外稳定性测定
采用放射性HPLC测定体外稳定性。取适量探针分别与PBS(pH 7.4)和FBS在37 ℃下孵育不同时间(0, 1, 2, 4, 6 h)。在每个时间点取样稀释至浓度为100 μCi/mL,取10 μL进样,利用放射性HPLC分析放射性化学纯度。HPLC条件为甲醇 ∶水 ∶三氟乙酸=40.0 ∶60.0 ∶0.1(V∶V∶V)。
1.5 探针[18F]7的脂水分配系数测定
取探针(1 μCi)加入到含有正辛醇(3.0 mL)和PBS(3.0 mL, pH 7.4)的离心管中,室温下涡旋振荡5 min,离心(4000 r/min,5 min)后分别取正辛醇(1.0 mL)和PBS(1.0 mL)于γ计数管中测定放射性计数(CPM),取1.0 mL正辛醇相加入到含正辛醇(2.0 mL)和PBS(3.0 mL)的离心管中。重复上述操作5次,根据公式LogP=Log(CPM正辛醇/CPMPBS)计算得到探针的脂水分配系数,结果用平均值±标准差表示。
2 结果与讨论
2.1 标记前体化合物10的化学结构分析
标记前体化合物10通过质谱和高效液相色谱进行初步表征,结果如图3所示,HPLC保留时间为16 min,纯度大于99%,质谱峰分子量514.29对应{[M+H]+}的分子离子峰。通过1H NMR和13C NMR进一步证明了标记前体化合物10的结构,结果如图4~5所示。
2.2 探针[18F]7的放射性化学分析
采用前体化合物10进行一步法18F标记,经过半制备和C18纯化得到探针[18F]7。探针[18F]7与化合物7共进样,结果如图7所示,探针[18F]7的保留时间与非放射性对照化合物7保留时间一致且探针结构正确,放射性化学性纯度大于99%,放射性化学产率为5%。该标记方法可应用于市售的18F多功能合成模块进行一步法自动化标记,有利于临床上放射性药物的生产和应用。

Time/min
2.3 探针[18F]7的体外稳定性
良好的探针体外稳定性有利于体内生物学活性的研究,探针[18F]7体外稳定性结果如图8所示,探针[18F]7在PBS和FBS中6 h内仅有1个明显的色谱主产品峰,未见其它放射性杂质,表明探针[18F]7在PBS和FBS中有较好的稳定性,有利于探针进一步的体内生物学研究。

Time/min
2.4 探针[18F]7的脂水分配系数
脑显像探针的亲脂性是保证药物透过血脑屏障的重要因素之一,研究表明,脂水分配系数系数在1.0~3.5之间[15],易于透过血脑屏障。本文研制的探针[18F]7的LogP值为2.15±0.17,表明探针[18F]7亲酯性较好,有透过血脑屏障的潜力。
本文采用氘代合成策略实现了一种DAT探针[18F]FP-CIT的氘代结构[18F]FP-CIT-d6([18F]7)的合成。首先合成非放射性对照化合物7和标记前体化合物10,再利用18F亲核取代法标记制备了高纯度的放射性探针[18F]7。体外理化性质测定结果表明:探针具有较好的体外稳定性和亲酯性。简便的标记方法和良好的理化性质,为探针[18F]7进一步的体内生物活性研究提供了坚实的基础。
猜你喜欢
农村青少年科学探究(2022年4期)2022-07-29
天津医科大学学报(2021年1期)2021-12-05
云南化工(2020年11期)2021-01-14
应用化工(2020年9期)2020-09-29
现代检验医学杂志(2016年5期)2016-08-20
环境与生活(2016年6期)2016-02-27
太空探索(2015年10期)2015-07-18
中南民族大学学报(自然科学版)(2014年4期)2014-08-06
茶叶通讯(2014年2期)2014-02-27
城市道桥与防洪(2014年3期)2014-01-08
