
铜催化串联反应一锅合成吲哚螺四氢喹啉酮衍生物
2023-08-19 00:30钟雪,何菱
合成化学 2023年8期
钟 雪, 何 菱
(四川大学 华西药学院,四川 成都 610041)
吲哚螺环化合物是多种生物活性天然产物和人工合成的小分子化合物的重要结构单元,由于吲哚衍生物与单环或多环形成非平面的螺环结构的特殊性,表现在与其生物靶点的三维空间作用时具有良好的适应性,因此具有较大的成药潜力[5-7]。已有研究报道该类化合物具有抗肿瘤、抗炎、抗菌和抗病毒等活性[1-9],其中,吲哚螺四氢喹啉及其结构类似物就是其中不可或缺的一类。如西帕加明(NITD609)是治疗疟疾的先导化合物,具有类似螺环吲哚结构的Surugatoxin是烟碱型乙酰胆碱受体的神经节阻滞剂。由于该类化合物在生物活性方面的广泛应用,人们对其合成策略和生物活性的开发不断发展。迄今为止,合成吲哚螺四氢喹啉的方法主要有[3+3]环加成,Michael/Michael串联,氮杂Diels-Alder反应和Povarov反应等[10-14]。
虽然吲哚螺氮杂六元环的合成方法较多,但对于其中的吲哚螺四氢喹啉的合成策略却很有限,为便于该类母核化合物多样性的发展和开展相应衍生物生物活性的研究,继续开发吲哚螺四氢喹啉类化合物的合成方法是十分必要的。
鉴于此,本文以硝基取代的吲哚烷酮衍生物为底物,乙酰丙酮钼和三氟甲烷磺酸铜为催化剂,三苯基膦为还原剂,甲苯为溶剂,以中等以上收率得到了4个吲哚螺四氢喹啉酮衍生物(图1),其结构经1H NMR,13C NMR和HR-MS(ESI)表征。
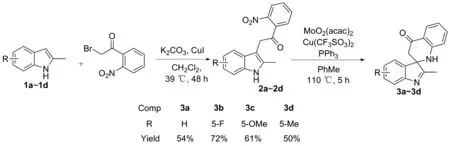
图1 吲哚螺四氢喹啉酮衍生物的合成路线
1 实验部分
1.1 仪器与试剂
Varian Mercury 400/600 MHz型核磁共振仪(DMSO-d6, CDCl3为溶剂,TMS为内标);Bruker DaltonicsData analysis 3.2 mass spectrometer型质谱仪。
所用试剂均为分析纯,未加以特别说明时均为直接使用,部分溶剂的干燥处理按照化学Vogle’s手册进行。
1.2 合成
(1)2a~2d的合成通法
将吲哚衍生物1a~1d(1.0 eq)置于干燥的反应瓶中,加入无水CH2Cl2溶解。然后将溴代邻硝基苯乙酮(1.2 eq),炒制的K2CO3(6.0 eq)与CuI(0.1 eq)一并加入反应体系中。TLC监测,反应48 h结束,加入饱和碳酸铵淬灭反应,CH2Cl2萃取,饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压浓缩,柱色谱分离(洗脱剂:石油醚/乙酸乙酯=8/1,V/V),得棕色固体2a~2d。
2-(2-甲基-1H-吲哚-3-基)-1-(2-硝基苯基)乙-1-酮(2a):棕色固体,收率55%;1H NMR(400 MHz, CDCl3)δ: 8.07(dd,J=7.5 Hz, 1.8 Hz, 1H), 7.93(s, 1H), 7.48(tt,J=7.6 Hz, 3.6 Hz, 2H), 7.35(d,J=7.7 Hz, 1H), 7.21(d,J=7.9 Hz, 1H), 7.11~7.01(m, 3H), 4.16(s, 2H), 2.24(s, 3H);13C NMR(150 MHz, DMSO-d6)δ: 200.2, 146.2, 136.7, 135.6, 134.6, 134.5, 131.7, 128.8, 128.8, 124.6, 120.6, 118.8, 118.0, 110.8, 102.4, 38.5, 11.7; HR-MS(ESI) calcd for C17H14N2NaO3{[M+Na]+}317.1004, found 317.0897。
2-(5-氟-2-甲基-1H-吲哚-3-基)-1-(2-硝基苯基)乙-1-酮(2b):棕色固体,收率53%;1H NMR(400 MHz, DMSO-d6)δ: 11.01(s, 1H), 8.16~7.99(m, 1H), 7.88~7.64(m, 3H), 7.63 ~7.30(m, 1H), 7.25~7.04(m, 1H), 6.86~6.68(m, 1H), 4.23(s, 2H), 2.26(s, 3H);13C NMR(150 MHz, DMSO-d6)δ: 200.1, 158.0(d,J=79.5 Hz), 146.2, 136.8(d,J=6.0 Hz), 134.7, 132.2, 131.7, 129.3(d,J=10.5 Hz), 128.7, 125.6, 124.6, 111.6(d,J=10.5Hz), 108.3(d,J=25.5Hz), 103.1, 102.9, 38.3, 11.8; HR-MS(ESI) calcd for C17H13FN2NaO3{[M+Na]+}335.0910, found 335.0802。
2-(5-甲氧基-2-甲基-1H-吲哚-3-基)-1-(2-硝基苯基)乙-1-酮(2c):棕色固体,收率57%;1H NMR(400 MHz, DMSO-d6)δ: 10.72(s, 1H), 8.07(d,J=8.5 Hz, 1H), 7.79(t,J=7.4 Hz, 1H), 7.69(ddd,J=7.7 Hz, 5.6 Hz, 1.7 Hz, 2H), 7.09(d,J=8.7 Hz, 1H), 6.86(d,J=2.4 Hz, 1H), 6.60(dd,J=8.8 Hz, 2.4 Hz, 1H), 4.20(s, 2H), 3.70(s, 3H), 2.22(s, 3H);13C NMR(150 MHz, DMSO-d6)δ: 200.2, 153.5, 146.3, 136.6, 135.2, 134.5, 131.7, 130.6, 129.2, 128.9, 124.6, 111.4, 110.2, 102.3, 100.4, 55.7, 38.4, 11.8; HR-MS(ESI) calcd for C18H17N2O4{[M+H]+}325.1110, found 325.1240。
2-(2,5-二甲基-1H-吲哚-3-基)-1-(2-硝基苯基)乙-1-酮(2d):浅棕色固体,收率61%;1H NMR(600 MHz, CDCl3)δ: 8.09(dd,J=7.8 Hz, 1.5 Hz, 1H), 7.79(s, 1H), 7.54~7.47(m, 2H), 7.14(s, 1H), 7.11(d,J=8.2 Hz, 1H), 7.08(dd,J=7.1 Hz, 1.8 Hz, 1H), 6.92(dd,J=8.1 Hz, 1.5 Hz, 1H), 4.15(s, 2H), 2.40(s, 3H), 2.21(s, 3H);13C NMR(150 MHz, Methanol-d4)δ: 193.6, 137.8, 131.7, 129.4, 125.9, 122.4, 120.4, 119.8, 115.9, 114.1, 111.3, 109.2, 104.3, 102.0, 93.7, 30.7, 16.2, 12.6; HR-MS(ESI) calcd for C18H16N2NaO3{[M+Na]+}331.1161, found 331.1053。
(2)3a~3d的合成通法
将2a~2d(1.0 eq)加入反应管中,随后依次加入三苯基膦(3.0 eq),乙酰丙酮钼(0.1 eq),三氟甲烷磺酸酮(0.1 eq),注入干燥的甲苯,氮气保护,无水操作。TLC监测反应,反应5 h,减压浓缩,柱色谱分离(洗脱剂:石油醚/乙酸乙酯=4/1~1/1,V/V),纯化得化合物3a~3d。
2-甲基-1′H-螺[吲哚-3,2′-喹啉]-4′(3′H)-酮(3a):淡红色固体,收率54%;1H NMR(600 MHz, DMSO-d6)δ: 7.67(dd,J=8.0 Hz, 1.7 Hz, 1H), 7.45(d,J=7.7 Hz, 1H), 7.39~7.34(m, 2H), 7.26~7.22(m, 2H), 7.11(t,J=7.4 Hz, 1H), 6.76(d,J=8.3 Hz, 1H), 6.73(t,J=7.5 Hz, 1H), 3.04(d,J=16.1 Hz, 1H), 2.55(d,J=16.1 Hz, 1H), 2.22(s, 3H);13C NMR(100 MHz, DMSO-d6)δ: 192.2, 182.8, 153.3, 151.5, 141.6, 136.2, 129.9, 126.5, 126.3, 122.2, 120.5, 117.9, 117.6, 116.8, 71.5, 43.1, 16.5; HR-MS(ESI) calcd for C17H15N2O{[M+H]+}263.1012, found 263.1179。
5-氟-2-甲基-1′H-螺[吲哚-3,2′-喹啉]-4′(3′H)-酮(3b):淡红色固体,收率72%;1H NMR(600 MHz, DMSO-d6)δ: 7.67(d,J=7.9 Hz, 1H), 7.46(dd,J=8.5 Hz, 4.6 Hz, 1H), 7.38(t,J=7.7 Hz, 1H), 7.29(s, 1H), 7.19(td,J=9.0 Hz, 2.6 Hz, 1H), 7.07(dd,J=8.0 Hz, 2.5 Hz, 1H), 6.76(d,J=8.3 Hz, 1H), 6.74(t,J=7.7 Hz, 1H), 2.96(d,J=16.2 Hz, 1H), 2.73(d,J=16.2 Hz, 1H), 2.18(s, 3H);13C NMR(150 MHz, DMSO-d6)δ: 191.9, 183.2(d,J=3 Hz), 161.6, 160.0, 151.2, 149.6, 143.5(d,J=7.5 Hz), 136.3, 126.5, 121.6(d,J=9.0 Hz), 117.8, 116.8, 116.3(d,J=24.0 Hz), 110.2(d,J=24.0 Hz), 71.8, 42.7, 16.6; HR-MS(ESI) calcd for C17H14FN2O{[M+H]+}281.1012, found 281.1012。
5-甲氧基-2-甲基-1′H-螺[吲哚-3,2′-喹啉]-4′(3′H)-酮(3c):淡红色固体,收率61%;1H NMR(600 MHz, DMSO-d6)δ: 7.66(dd,J=8.0 Hz, 1.6 Hz, 1H), 7.38~7.33(m, 2H), 7.25(s, 1H), 6.90(dd,J=8.4 Hz, 2.6 Hz, 1H), 6.85(d,J=2.6 Hz, 1H), 6.77(d,J=8.3 Hz, 1H), 6.72(t,J=7.5 Hz, 1H), 3.68(s, 3H), 2.92(d,J=16.2 Hz, 1H), 2.67(d,J=16.2 Hz, 1H), 2.15(s, 3H);13C NMR(150 MHz, DMSO-d6)δ: 192.2, 180.5, 158.1, 151.4, 146.8, 143.0, 136.2, 126.4, 120.8, 117.8, 117.6, 116.8, 113.6, 109.7, 71.5, 56.0, 43.2, 16.5; HR-MS(ESI) calcd for C18H17N2O2{[M+H]+}293.1212, found 293.3381。
2,5-二甲基-1′H-螺[吲哚-3,2′-喹啉]-4′(3′H)-酮(3d):淡红色固体,收率50%;1H NMR(400 MHz, DMSO-d6)δ: 7.67(dd,J=7.9 Hz, 1.6 Hz, 1H), 7.37(ddd,J=8.5 Hz, 7.0 Hz, 1.7 Hz, 1H), 7.32(d,J=7.8 Hz, 1H), 7.25(s, 1H), 7.15(dd,J=7.9 Hz, 1.6 Hz, 1H), 7.06(s, 1H), 6.76(d,J=8.3 Hz, 1H), 6.72(t,J=7.4 Hz, 1H), 3.01(d,J=16.1 Hz, 1H), 2.54(d,J=16.2 Hz, 1H), 2.22(s, 3H), 2.19(s, 3H);13C NMR(150 MHz, DMSO-d6)δ: 192.2, 181.8, 151.5, 151.2, 141.7, 136.2, 135.5, 130.2, 126.5, 122.8, 120.1, 117.8, 117.5, 116.7, 71.3, 43.1, 21.4, 16.4; HR-MS(ESI) calcd for C18H17N2O{[M+H]+}277.1263, found 277.1335。
2 结果与讨论
2.1 反应条件选择
以3b(其单晶结构如图2所示)的合成为例,考察了催化剂、溶剂、还原剂、反应温度和时间对反应的影响。
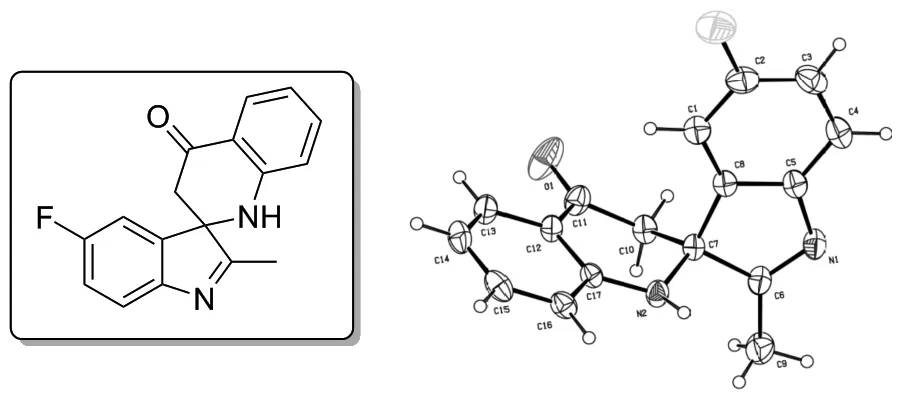
图2 化合物3b的单晶结构
(1) 催化剂
以三苯基膦为还原剂,乙酰丙酮钼为脱氧催化剂,干燥甲苯为反应溶剂,考察不同催化剂在不同温度下对反应的影响,其结果如表1所示。当温度分别为30 ℃、 60 ℃、 80 ℃、 100 ℃时,含金属钯、铜、银、镍、铬、镱和钨等的催化剂均使反应收率低于10%。当反应温度升为110 ℃时,三氟甲烷磺酸铜作催化剂的收率可高达72%。因此,选用乙酰丙酮钼与三氟甲烷磺酸铜组合作为硝基还原与氮宾插入串联反应催化剂,最后实现吲哚螺四氢喹啉衍生物的合成。
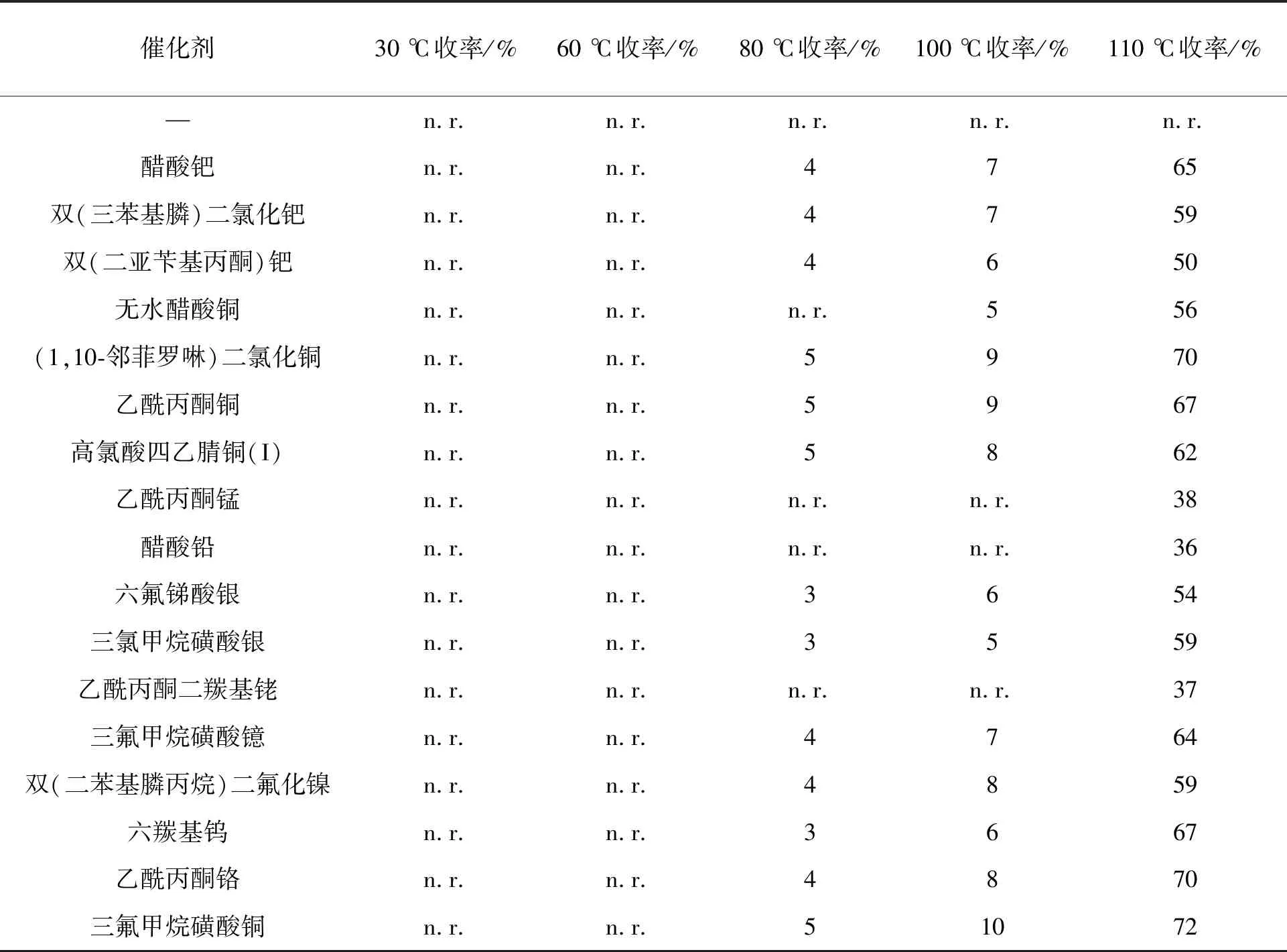
表1 不同温度下催化剂对反应收率的影响
(2) 溶剂
表2为溶剂的筛选。以乙酰丙酮钼和三氟甲烷磺酸铜为催化剂,三苯基膦为还原剂,溶剂均做无水处理。由表2可见,溶剂为二氯甲烷、四氢呋喃、丙酮、乙腈、N,N-二甲基甲酰胺、二甲基亚砜时,均未检测到产物生成,而在甲苯中收率为72%,因此选择无水甲苯作为反应溶剂。
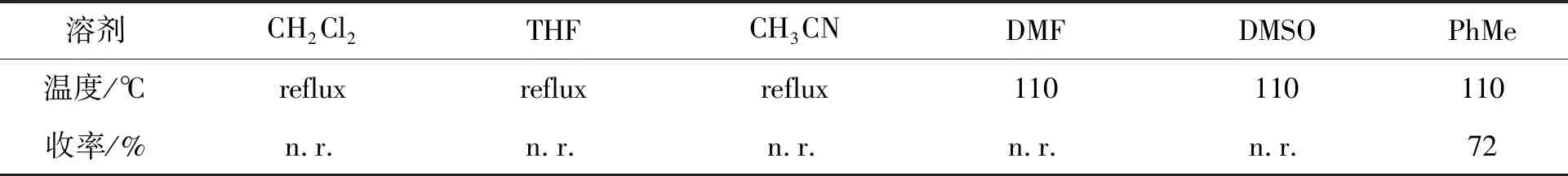
表2 溶剂对反应收率的影响
(3) 还原剂
表3为还原剂的筛选。以乙酰丙酮钼和三氟甲烷磺酸铜为催化剂,无水甲苯作溶剂,在110 ℃的条件下对不同还原剂进行筛选。由表3可见,不使用还原剂或使用还原剂三甲基膦,反应均不进行。还原剂为三苯基膦时,收率可达72%,而使用还原剂S-(-)-1,1′-联萘-2,2′-双二苯膦和4,5-双二苯基膦-9,9-二甲基氧杂蒽时,收率分别为43%和51%,因此选择三苯基膦为还原剂。

表3 还原剂对反应收率的影响
(4) 反应温度和反应时间
表4为反应温度和反应时间的筛选。以乙酰丙酮钼和三氟甲烷磺酸铜为催化剂,三苯基膦为还原剂,无水甲苯为溶剂,筛选不同反应温度和时间对反应的影响。由表4可见,反应在30 ℃、 60 ℃时均不发生,然而在80 ℃和100 ℃时,反应发生,但收率也低于10%。当温度升高至110 ℃时,收率随反应时间的增长而增大,反应时间为12 h时,收率为72%。在反应时间为12 h,收率随反应温度(120 ℃, 130 ℃和140 ℃)的升高依次下降。因此,反应的最佳温度为110 ℃,且在此温度下的最佳反应时间为12 h。
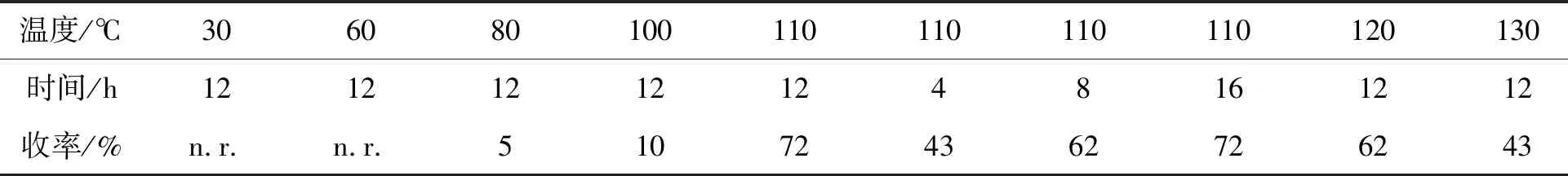
表4 温度和时间对反应的影响
综上,最终选择反应条件为: 乙酰丙酮钼和三氟甲烷磺酸铜为催化剂,三苯基膦为还原剂,甲苯为溶剂,于110 ℃反应12 h,3b收率为72%。并通过单晶培养,用单晶X-射线衍射确定了产物单晶结构。
2.2 反应机理的推测
结合文献[15],推测反应的过程如下(图3),硝基首先被乙酰丙酮钼和三苯基膦还原为氮宾中间体c;接着,c与三氟甲烷磺酸铜形成N—Cu键,并对活泼的吲哚3位进行插入,形成C—N键,然后与吲哚双键碳和吲哚氮上的氢形成六元中间体d;吲哚氮氢键断裂,并同时双键重排形成中间体e;最后三氟甲烷磺酸铜离去,吲哚重排成亚胺,生成吲哚螺四氢喹啉酮化合物3a。
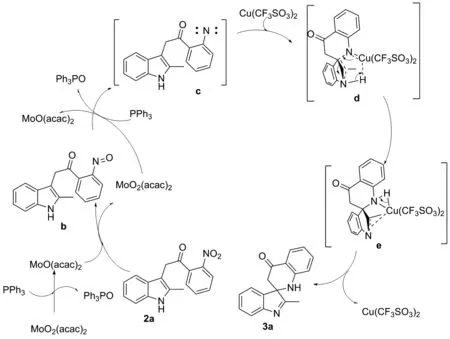
图3 可能的反应途径
本文以硝基取代的吲哚烷酮衍生物为底物,乙酰丙酮钼和三氟甲烷磺酸铜为催化剂,三苯基膦为还原剂,无水甲苯为溶剂,将吲哚烷酮底物中的硝基进行还原,而后氮宾插入,分子内环化构建了吲哚螺四氢喹啉酮化合物,最终以中等以上收率得到了4个吲哚螺四氢喹啉酮衍生物。该研究不仅拓宽了吲哚螺环类化合物的合成方法,还为该类化合物的后续活性研究奠定了基础。
猜你喜欢
能源工程(2021年1期)2021-04-13
中学化学(2019年4期)2019-08-06
中学化学(2019年4期)2019-08-06
中成药(2017年7期)2017-11-22
国外医药(抗生素分册)(2016年1期)2016-07-10
合成化学(2015年1期)2016-01-17
中国生化药物杂志(2015年4期)2015-07-07
湖南师范大学自然科学学报(2015年2期)2015-02-27
金属加工(热加工)(2014年23期)2014-11-25
安徽工业大学学报(自然科学版)(2014年4期)2014-07-11
