
鹅去氧胆酸关键中间体的合成工艺研究
2023-08-19 00:30:04王盈盈
合成化学 2023年8期
王盈盈, 孙 彬,2, 金 灿,2*
(1. 浙江工业大学 药学院 长三角绿色制药协同创新中心,浙江 杭州 310014; 2. 国家化学原料药合成工程技术研究中心,浙江 杭州 310014)
鹅去氧胆酸(Chenodeoxycholic acid, CDCA)在临床上被广泛用于治疗胆结石和其他肝胆疾病[1]。研究表明,CDCA在抗炎、降血压、解痉、平喘及调节免疫等方面具有一定的药理活性[2-3]。
此外,鹅去氧胆酸也是合成熊去氧胆酸、奥贝胆酸及其他甾体化合物的关键原料[4-6]。而3α-羟基-6-烯胆烷酸甲酯(1)是合成CDCA的关键中间体,即在C-6/7位形成双键,双键经环氧化生成环氧,最后开环选择性合成7α-羟基。王钟麒[7]、张飞[8]和刘明等[9]均采取此种路线合成CDCA,其中刘明等[9]采用Shapiro反应,在温和的条件下将酮基转变成烯烃类化合物;张国明[10]、卢茂芳[11]和张宗磊等[12]以CDCA为原料,通过将7α-羟基氧化还原即可合成熊去氧胆酸。根据IIDA[13]和KANG等[14]的研究可知,3α-羟基-6-烯胆烷酸甲酯的3-OH经保护后,通过环氧开环和水解等一系列步骤可合成得到α-鼠胆酸。3α-羟基-6-烯胆烷酸甲酯作为重要中间体具有广阔的应用前景,因此开发绿色、简便和高效的3α-羟基-6-烯胆烷酸甲酯合成方法成为制药工作者近年研究的热点。
合成3α-羟基-6-烯胆烷酸甲酯的关键步骤是氧化猪去氧胆酸的C-6羟基得到中间体3α-羟基-6-氧代-5β-胆酸甲酯(2)。猪去氧胆酸的C-3位和C-6位均为仲羟基,传统方法采用直接选择性氧化C-6羟基,然而此方法存在氧化选择性差的问题。周维善[15]、蒋忠良等[16]以重铬酸吡啶盐为氧化剂氧化C-6羟基合成化合物2,收率分别为62.00%和48.20%,但含铬废水量大,后处理繁琐;窦倩[17]、李阳等[18]采用N-溴代琥珀酰亚胺(NBS)作为选择性氧化剂,但同样存在氧化反应选择性问题。2018年,韩迎等[19]利用Jones试剂作为氧化剂,在-20 ℃条件下以67.00%收率得到化合物2,但该方法需要低温,条件苛刻,不利于工业化生产;2020年,LIANG等[20]以2-碘酰苯甲酸(IBX)为氧化剂,以叔丁醇为溶剂,在85 ℃下以72.00%收率合成得到化合物2,但IBX在有机溶剂中溶解性不好,且高价碘试剂成本高,性质不稳定。总体来看,上述报道的方法均需消耗化学当量级的氧化剂,不仅原子利用率低,产生三废多,污染环境,而且反应选择性较差,易产生C-3羟基氧化产物及双氧化产物,不易分离提纯。
本文以猪去氧胆酸为起始原料(图1),先经酯化保护其侧链的羧基得到甲酯化合物3。然后选择大位阻保护基叔丁基二甲基氯硅烷(tert-Butyldimethylsilyl chloride, TBMDSCl)实现对C-3羟基选择性保护,以91.60%收率合成化合物4;再采用Fe(NO3)3/4-OH-TEMPO/NaCl体系[21]催化氧气氧化C-6羟基,以92.80%收率得到中间体酮5。利用四丁基氟化铵脱去硅醚保护基合成化合物2(收率94.06%)后,不经分离纯化,直接与对甲苯磺酰肼进行腙化反应合成中间体腙6。随后进行Shapiro反应以合成C-6/7位烯烃产物3α-羟基-6-烯胆烷酸甲酯(1),收率为77.32%。
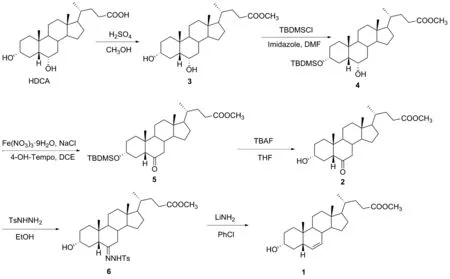
图1 3α-羟基-6-烯胆烷酸甲酯的合成路线
对比之前报道的方法,本研究通过先对猪去氧胆酸甲酯的C-3羟基选择性保护,再以氧气为氧化剂,催化量的硝酸铁、4-OH-TEMPO和NaCl为催化剂氧化C-6羟基,然后脱硅醚保护合成3α-羟基-6-氧代-5β-胆酸甲酯。其中,氧化步骤采用天然易得的空气作为氧化剂,且4步总收率可达78.36%。最后通过腙化、脱腙合成鹅去氧胆酸关键中间体3α-羟基-6-烯胆烷酸甲酯。该路线总收率达55.21%。各步骤产物结构经1H NMR,13C NMR和HR-MS确证。
1 实验部分
1.1 仪器与试剂
Buchi型数字熔点仪;Bruker 400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Thermo Finnigan型质谱仪。
猪去氧胆酸,上海麦克林生化科技有限公司。其余所用试剂均为分析纯。
1.2 合成
(1)3的合成
将猪去氧胆酸(10.00 g, 25.50 mmol)溶于甲醇(100.00 mL),加入浓H2SO4(0.20 mL, 3.80 mmol),室温反应24 h。反应结束,减压蒸馏除去甲醇,加入乙酸乙酯(100.00 mL)溶解,依次用饱和NaHCO3水溶液(3×20 mL)、饱和NaCl水溶液(3×20 mL)洗涤,收集乙酸乙酯层,加入无水MgSO4干燥,抽滤得滤液,减压蒸馏除去乙酸乙酯得到10.10 g白色固体化合物3,收率98.00%, m.p.146~148 ℃;1H NMR(400 MHz, CDCl3)δ: 4.09(dt,J=12.3 Hz, 4.7 Hz, 1H), 3.70(s, 3H), 3.68~3.61(m, 1H), 2.39(td,J=10.1 Hz, 5.0 Hz, 1H), 2.26(ddd,J=15.6 Hz, 9.6 Hz, 6.5 Hz, 1H), 0.96(d, 3H ), 0.92(s, 3H), 0.68(s, 3H);13C NMR(101 MHz, CDCl3)δ: 174.72, 71.67, 68.15, 56.21, 51.45, 50.46, 42.69, 41.51, 39.82, 39.65, 39.44, 35.36, 35.34, 35.05, 34.62, 32.87, 31.02, 31.00, 30.63, 28.13, 23.69, 22.77, 20.59, 18.27, 11.76; HR-MS(ESI)m/z: Calcd for C25H43O4{[M+H]+}407.3083, found 407.3079。
(2)4的合成
向100 mL圆底烧瓶中依次加入化合物3(10.00 g, 24.63 mmol)、N,N-二甲基甲酰胺(DMF)(30.00 mL),室温搅拌,待溶解完全后,依次加入TBDMSCl(5.40 g, 35.83 mmol)、咪唑(4.90 g,71.95 mmol),室温反应3 h,TLC跟踪反应进程。反应结束后,加入乙酸乙酯,用水萃取3次,分离得有机层,无水硫酸钠干燥,抽滤得滤液,减压蒸馏回收乙酸乙酯,甲醇重结晶,得到白色固体11.73 g,收率91.60%, m.p.149~152 ℃;1H NMR(400 MHz, CDCl3)δ: 4.02(dt,J=11.8 Hz, 4.7 Hz, 1H), 3.68(s, 3H), 3.60(dt,J=10.8 Hz, 6.0 Hz, 1H), 2.37(ddd,J=15.3 Hz, 10.2 Hz, 5.1 Hz, 1H), 2.23(ddd,J=15.6 Hz, 9.6 Hz, 6.4 Hz, 1H), 0.94(s, 3H), 0.92(d,J=3.9 Hz, 3H), 0.89(s, 9H), 0.66(s, 3H), 0.05(s, 6H);13C NMR(101 MHz, CDCl3)δ: 174.65, 72.53, 68.21, 56.06, 55.96, 51.40, 48.56, 42.85, 39.92, 39.66, 35.95, 35.80, 35.32, 35.12, 34.90, 31.06, 30.98, 30.94, 29.47, 28.09, 25.94(3C), 24.18, 23.46, 20.74, 18.24, 11.99, -4.58, -4.60; HR-MS(ESI)m/z: Calcd for C31H57O4Si{[M+H]+}521.3947, found 521.3951。
(3)5的合成
在氧气氛围下,依次向50 mL圆底烧瓶中加入Fe(NO3)3·9H2O(0.25 mmol, 0.10 g)、 4-OH-TEMPO(0.52 mmol, 0.09 g)、化合物4(5.00 mmol, 2.60 g)、 NaCl(0.51 mmol, 0.03 g)和1,2-二氯乙烷(20.00 mL),在25 ℃下搅拌,TLC跟踪反应进程。反应结束后,反应体系经水洗、二氯甲烷萃取后分液得有机层,无水硫酸钠干燥,抽滤得滤液,减压蒸馏浓缩除去溶剂,得到粗产品。粗产品经柱层析(洗脱剂:乙酸乙酯/石油醚=1/15,V/V)纯化,得到2.40 g白色固体化合物5,收率92.80%, m.p.141~145 ℃;1H NMR(400 MHz, CDCl3)δ: 3.68(d,J=1.4 Hz, 3H), 3.58(tt,J=10.3 Hz, 4.8 Hz, 1H), 2.37(ddd,J=15.3 Hz, 10.0 Hz, 5.1 Hz, 1H), 2.25(dt,J=8.6 Hz, 6.4 Hz, 1H), 2.23~2.16(m, 2H), 2.11(dd,J=12.9 Hz, 4.5 Hz, 1H), 0.94(d,J=6.5 Hz, 3H), 0.89(s, 9H), 0.84(s, 3H), 0.66(s, 3H), 0.06(s, 3H), 0.05(s, 3H);13C NMR(101 MHz, CDCl3)δ: 213.88, 174.60, 71.17, 59.82, 56.76, 55.86, 51.46, 43.11, 42.86, 39.73, 39.60, 37.91, 37.10, 35.50, 35.28, 34.67, 31.06, 30.94, 30.56, 27.98, 25.85, 23.96, 23.23, 20.83, 18.24, 18.17, 11.95, -4.68, -4.71; HR-MS(ESI)m/z: Calcd for C31H55O4Si{[M+H]+}519.3755, found 519.3761。
(4)2的合成
将化合物5(1.00 mmol, 0.52 g)、四丁基氟化铵水合物(1.50 mmol, 0.39 g)、四氢呋喃(THF)(5.00 mL)加入到50 mL圆底烧瓶中,室温搅拌1 h,TLC跟踪反应进程。反应结束后,加入乙酸乙酯(30 mL)和饱和NaCl水溶液(3×20 mL)洗涤。分离得有机层,用无水硫酸钠干燥,抽滤得滤液,减压蒸馏浓缩除去有机溶剂,异丙醇重结晶得0.38 g白色固体化合物2,收率94.06%, m.p.134~136 ℃;1H NMR(600 MHz, CDCl3)δ: 3.66(s, 3H), 3.65~3.60(m, 1H), 2.36(ddd,J=15.4 Hz, 10.1 Hz, 5.2 Hz, 1H), 2.26~2.21(m, 1H), 2.20~2.16(m, 2H), 2.15~2.09(m, 1H), 0.93(d,J=6.5 Hz, 3H), 0.84(s, 3H), 0.65(s, 3H);13C NMR(151 MHz, CDCl3)δ: 213.96, 174.68, 70.09, 59.42, 56.81, 55.79, 51.52, 43.09, 42.90, 39.98, 39.59, 37.97, 37.06, 35.27, 34.86, 34.38, 31.04, 30.90, 29.82, 27.98, 23.96, 23.16, 20.83, 18.23, 11.95; HR-MS(ESI)m/z: Calcd for C25H41O4{[M+H]+}405.2975, found 405.2979。
(5)6的合成
将化合物2(0.94 mmol, 0.38 g)置于25 mL圆底烧瓶,加入对甲苯磺酰肼(1.09 mmol, 0.21 g)、对甲苯磺酸(0.06 g, 0.35 mmol)和无水乙醇(5.00 mL),室温搅拌3 h,TLC跟踪反应进程。待反应结束后,加入NaHCO3水溶液,调节pH值至7,加入乙酸乙酯(30 mL)和饱和NaCl水溶液(3×20 mL)洗涤,分离得有机层,用无水硫酸钠干燥,抽滤得滤液,减压蒸馏浓缩除去溶剂。粗产品经柱层析(洗脱剂:乙酸乙酯/石油醚=1/5,V/V)纯化,得到0.49 g白色固体化合物6,收率91.13%, m.p.95~97 ℃;1H NMR(400 MHz, DMSO-d6)δ: 7.70(dd,J=8.2 Hz, 4.0 Hz, 2H), 7.37(d,J=7.9 Hz, 2H), 3.58(s, 3H), 3.45~3.33(m, 1H), 2.73~2.60(m, 1H), 2.37(s, 3H), 2.36~2.27(m, 1H), 2.22(td,J=10.4 Hz, 10.0 Hz, 5.9 Hz, 1H), 0.94(d,J=6.4 Hz, 3H), 0.84(s, 3H), 0.66(s, 3H);13C NMR(101 MHz, DMSO-d6)δ: 174.21, 163.73, 143.28, 136.98, 129.72, 127.79, 69.21, 56.47, 55.74, 52.27, 51.67, 43.02, 42.88, 36.70, 36.60, 35.59, 35.19, 34.50, 31.03, 30.78, 30.17, 28.76, 28.10, 24.13, 22.88, 22.83, 21.47, 18.53, 12.26, 12.17; HR-MS(ESI)m/z: Calcd for C32H49N2O5S{[M+H]+}572.8055, found 572.8049。
(6)1的合成
将氨基锂(1.75 mmol,0.04 g)溶于5.00 mL氯苯,室温搅拌溶解,待用。在氮气氛围下,向上述溶液中滴加腙化物6的氯苯溶液(0.50 mmol, 0.29 g, 5.00 mL PhCl),滴加完毕后,在回流温度下搅拌3 h,TLC跟踪反应进程。反应结束后,冷却至室温,调节pH至中性。加入和饱和NaCl水溶液(3×20 mL)洗涤。分离得有机层,用无水硫酸钠干燥,抽滤得滤液,减压蒸馏浓缩除去溶剂。粗产品经柱层析(洗脱剂:乙酸乙酯/石油醚=1/3,V/V)纯化得到0.15 g白色固体化合物1,收率77.32%, m.p.101~103 ℃;1H NMR(400 MHz, CDCl3)δ: 5.54(d,J=9.7 Hz, 1H), 5.46(d,J=10.1 Hz, 1H), 3.69(s, 3H), 3.60(s, 1H), 2.36(dd,J=10.7 Hz, 5.0 Hz, 1H), 2.25(d,J=7.9 Hz, 1H), 0.95(d,J=6.4 Hz, 3H), 0.87(s, 3H), 0.71(s, 3H);13C NMR(101 MHz, CDCl3)δ: 174.78, 130.64, 128.11, 71.22, 55.77, 54.72, 51.52, 43.89, 43.36, 40.12, 39.78, 37.64, 35.37, 34.39, 33.24, 31.08, 31.01, 30.45, 29.71, 28.28, 23.90, 22.71, 20.55, 18.27, 12.05; HR-MS(ESI)m/z: Calcd for C25H41O3{[M+H]+}389.5911, found 389.5916。
3α-羟基-6-氧代-5β-胆酸甲酯是合成3α-羟基-6-烯胆烷酸甲酯的关键中间体。通过实验研究发现,硅醚类保护基对猪去氧胆酸3-OH有特殊的选择性,可以高选择性地保护3-OH,而6-OH基本不受影响。本文通过用大位阻硅醚保护基保护3-OH,再对6-OH进行氧化,避免了传统氧化方法中3-OH同时被氧化的情况,提高了氧化选择性。此外,本文重点考察了4和5的合成工艺条件。
2.1 硅醚保护反应条件的优化
为优化4的合成方法,研究了TBMDSCl用量、溶剂种类和反应温度对4的收率的影响。由表1可知,TBMDSCl的用量对该反应影响较大,实验过程中发现,随着TBMDSCl用量增加,4收率逐渐提高,当TBMDSCl的用量为1.5 eq.时,收率最高(Entry 1~3)。但再增加TBMDSCl的用量,6-OH也会被硅醚保护,生成部分双保护副产物。当采用CH2Cl2、 CHCl3、 DMF和THF作为反应溶剂时,发现DMF的反应效果最好(Entry 5)。同时还考察了温度对反应收率的影响。分别设置了-10 ℃、 0 ℃、 25 ℃和45 ℃ 4个梯度(Entry 5, 7~9),研究表明,随着温度升高,4收率不断提高。当反应温度为25 ℃时,收率最高为94.34%。继续升高温度,会导致双保护副产物增加,4收率降低。综上所述,合成4的最优工艺条件为:以DMF为溶剂,TBMDSCl用量为1.5 eq.,咪唑用量为3.0 eq., 25 ℃下反应3 h。

表1 4的合成条件优化
2.2 氧化合成5的条件优化
本文以氧气为氧化剂,采用Fe(NO3)3/4-OH-TEMPO/NaCl催化体系氧化6-OH,从而合成5。由表2可知,在空气氛围下(Entry 1)反应收率远低于在氧气氛围下的反应收率,因此氧气为促进该反应不可或缺的条件。分别采用ClCH2CH2Cl、 CH2Cl2和EtOAc作为反应溶剂,结果表明,ClCH2CH2Cl的反应效果最好。进一步考察催化剂4-OH-TEMPO、 Fe(NO3)3·9H2O和NaCl的用量(Entry 2,5~8)可知,当4-OH-TEMPO、Fe(NO3)3·9H2O和NaCl的用量分别为10%(物质的量分数,下同)、5%和10%时,收率最高为95.17%,说明NaCl有加速反应的效果。由Entry 9可知,采用TEMPO和用4-OH-TEMPO效果相差不大,但4-OH-TEMPO价格更便宜。继续考察了温度对反应收率的影响,由Entry 10~11可知,降低温度至10 ℃时反应速率明显减慢,升高温度至40 ℃反应过程中杂质增多,导致反应收率降低。综上所述,合成5的最优工艺条件为:在氧气保护下,以ClCH2CH2Cl为溶剂,4-OH-TEMPO用量为10 %, Fe(NO3)3·9H2O用量为5%, NaCl用量为10 %,在25 ℃下反应20 h。

表2 5的合成条件优化
本研究报道了以猪去氧胆酸为起始原料,经甲酯化、3-OH硅醚保护、6-OH氧化、脱保护、腙化和脱腙6步合成得到鹅去氧胆酸关键中间体3α-羟基-6-烯胆烷酸甲酯,总收率为55.21%。该路线避免了当量级的重金属氧化试剂使用,具有氧化选择性高、原子经济性高和总收率较高等优点,为工业化生产提供了借鉴。
猜你喜欢
理化检验-化学分册(2021年10期)2021-11-29 14:50:42
云南化工(2020年11期)2021-01-14 00:50:52
中国塑料(2015年6期)2015-11-13 03:03:11
化工进展(2015年3期)2015-11-11 09:07:41
中国医药科学(2015年5期)2015-08-01 14:14:51
中国当代医药(2015年10期)2015-03-01 02:02:39
中国当代医药(2015年9期)2015-03-01 02:02:12
海军医学杂志(2015年2期)2015-02-27 13:47:35
中成药(2014年4期)2014-04-01 08:43:42
湖北科技学院学报(医学版)(2014年3期)2014-02-28 19:42:51
