
肾小管间质性肾病基因-表型关联性分析
2023-06-26 03:04杨雨蒙周雅丽杨单植邢国兰
郑州大学学报(医学版) 2023年3期
杨雨蒙,张 颖,魏 敏,周雅丽,杨单植,邢国兰
郑州大学第一附属医院肾脏内科 郑州 450052
肾小管间质性肾病是指病理损伤主要累及肾间质和肾小管的一组疾病,常伴有间质水肿、纤维化和肾小管萎缩[1]。根据病程长短,肾小管间质性肾病分为急性和慢性。急性肾小管间质性肾病是急性肾损伤的常见原因,占肾活检中急性肾损伤患者的18%~27%[2]。急性肾小管间质性肾病与炎症细胞介导的肾间质浸润有关,可发展为间质纤维化,从而转化为慢性肾小管间质性肾病[3]。肾小管间质性肾病可以由多种因素引起,最常见的病因是药物诱导[4],其次为自身免疫病、感染等因素。某些遗传因素也可导致肾小管间质性肾病,主要有常染色体隐性遗传和常染色体显性遗传两种遗传方式,前者主要指肾消耗病(NPHP),后者指常染色体显性遗传性肾小管间质性肾病(ADTKD)。NPHP已鉴定出超过20种不同的致病基因。NPHP1基因突变是导致NPHP最常见的形式,占所有病例的20%[5]。而ADTKD是一组由尿调素(UMOD)、黏蛋白1、肝细胞核因子-1b、肾素[6]和运输蛋白SEC61亚基α基因突变[7]引起的遗传疾病。遗传相关肾小管间质性肾病起病隐匿,临床表现无明显特异性,容易漏诊及误诊。因此对于疑似遗传相关肾小管间质性肾病,早期进行基因检测对于病因诊断以及后期治疗都十分重要。改善全球肾脏病预后组织指南[8]指出应该使用基因组学来定义和分层管理慢性肾脏病,对临床评估为疑似遗传相关的肾脏病考虑进行基因检测,可以为其提供潜在的分子遗传学证据。如今全外显子组测序技术已得到广泛应用。本文分析了24例肾小管间质性肾病的全外显子组测序结果,总结其共有的罕见遗传变异基因,以期进一步提高对遗传相关肾小管间质性肾病的认识。
1 对象与方法
1.1 研究对象收集2012年1月1日至2020年12月31日经郑州大学第一附属医院肾穿刺活检病理检查确诊为肾小管间质性肾病且年龄≤40岁的患者。排除标准:①合并其他原发性肾小球疾病、继发性肾小球疾病及遗传性肾小球疾病。②药物性、感染性、自身免疫性、代谢性、梗阻或反流引起的肾小管疾病。③临床资料不全以及失访者。本研究是根据赫尔辛基宣言进行的,并得到了该院伦理委员会审批(批号:2022-KY-0028-002),且所有研究对象均签署知情同意书。
1.2 研究方法
1.2.1临床指标 收集患者人口学资料及实验室检查结果,人口学资料包括性别、年龄等;实验室检查结果包括血尿素氮、血肌酐、血尿酸、尿密度、24 h尿蛋白量。医院病理系统中查阅病理报告,收集患者的肾组织病理检查结果。
1.2.2全外显子组测序 应用Tiangen DNA提取试剂盒提取患者入院时全血样本中的基因组DNA,采用Agilent SureSelect Human All Exon试剂盒进行建库和捕获实验,随后在Illumina HighSeq平台上进行测序。
1.2.3罕见潜在致病变异的筛选流程 应用ANNOVAR对变异进行注释,然后查阅数据库判断变异在人群中出现的频率[主要数据库:Freq_gnomAD_genome_ALL、Freq_esp6500siv2_all、Freq_1000g2015aug_all],基因的功能通过OMIM数据库进行分析。根据ACMG(2015)指南对基因突变进行致病性判断,去除意义不明、可能良性及良性的基因。选取总体样本10%以上患者共存的变异,应用Phenolyzer数据库进行基因-表型关联评分,筛选肾小管间质性肾病易感基因。进一步利用DAVID 6.8数据库对筛选出的基因进行GO分析和KEEG分析。应用String数据库(http://string-db.org)构建PPI网络图,并使用Cytoscape中的插件cytoHubba进行可视化分析,寻找枢纽基因。应用SWISS-Pdb Viewer软件分析基因突变前后编码蛋白三级结构的变化。
2 结果
2.1 临床表现及主要实验室检查结果本研究24例肾小管间质性肾病患者中,女9例,男15例;年龄18~40(26.17±8.82)岁。所有病例临床表现多种多样,但均出现不同程度的肾功能损害,血尿素氮升高至7.23~18.97(中位数11.70) mmol/L,血肌酐升高至129.0~464.0(中位数292.5) μmol/L,血尿酸升高至150.3~474.5(中位数276.5) μmol/L,而且有微量白蛋白尿[24 h尿蛋白定量为0.28~1.14(中位数0.79) g],肾小管间质损伤,浓缩能力下降,尿密度偏低[(1.01±0.01)×103kg/m3]。
2.2 肾小管间质性肾病的肾脏病理特征肾小球表现以缺血皱缩为主,共14例;其次为系膜轻度增生,共11例。肾小管病变以刷状缘脱落为主,共22例;其次为上皮细胞空泡、颗粒变性,均为21例;肾小管管腔扩张、细胞低平,共20例。肾间质以间质纤维化为主,共15例。
2.3 全外显子组测序结果24例患者均为散发病例,全外显子组测序检测出19例携带致病或可能致病的突变,主要包括UMOD、REN、CEP164、TMEM67、NPHP1、CEP290,与患者临床诊断疾病的遗传模式一致4例,其中3例为常染色体显性遗传,1例为常染色体隐性遗传。基因突变检测结果见表1。

表1 肾小管间质性肾病相关致病基因
2.4 与肾小管间质性肾病相关易感基因3例常染色体显性遗传肾小管间质性肾病患者共有61个罕见变异,包括非同义突变102个,根据基因-表型关联评分筛选肾小管间质性肾病易感基因(图1),共发现35个易感基因,其中高置信基因(分值>0.5)12个:VCP(1.701)、AKAP9(1.323)、LAMA5(1.075)、NCOR2(1.004)、TGFBR2(0.981)、BCAS3(0.963)、RBPJ(0.803)、PKD1(0.718)、PRKDC(0.691)、INHBB(0.637)、CLOCK(0.611)、GMDS(0.594)。
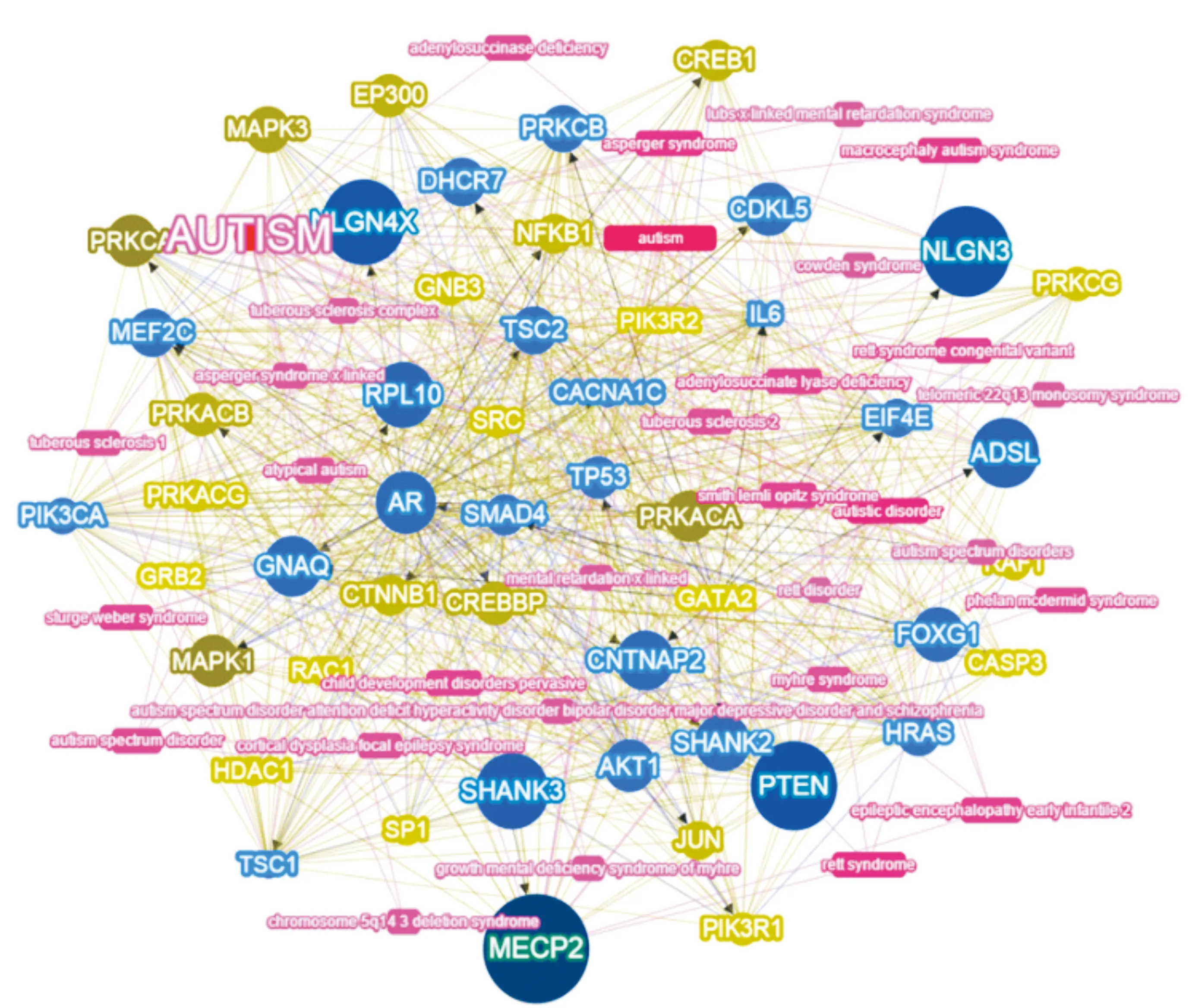
圆形节点代表基因,蓝色为报道过的基因,黄色为预测的基因;基因评分越高,颜色越深;矩形代表疾病,线段越长,颜色越深的贡献越大;蓝色线条代表两个基因之间的蛋白质-蛋自质相互作用;黄色线条代表两个基因之间的同一生物系统关系;绿色线条代表两个基因-基因-家族关系;黑色线条代表两个基因之间的转录相互作用,箭头方向表示从转录因子到目标基因
2.5 通路富集分析对12个肾小管间质性肾病易感基因进行GO分析,结果显示易感基因多富集在离子跨膜转运的调节、蛋白质转运的调控、蛋白质磷酸化等生物过程;富集在细胞质基质、离子通道、细胞内膜结合的细胞器等细胞组成;富集在蛋白质结合、转录因子结合、离子通道结合等分子功能。KEGG分析结果显示易感基因富集在TGF-β信号通路、细胞因子-细胞因子受体相互作用信号通路。见表2。
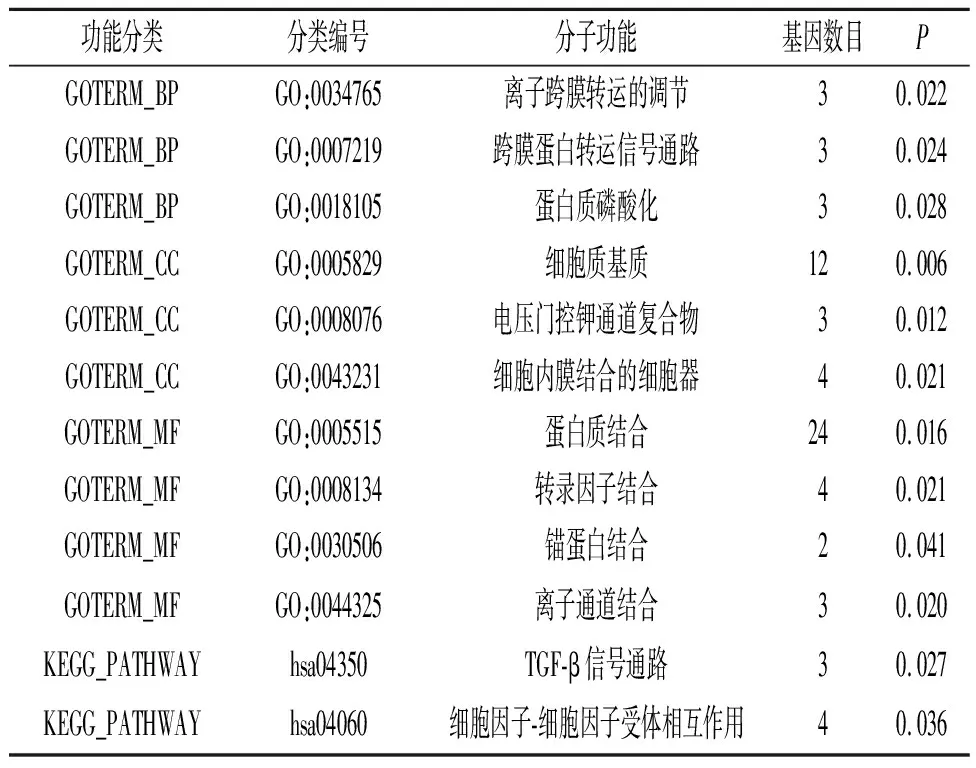
表2 易感基因通路富集分析结果
2.6 PPI网络图及枢纽基因的鉴定使用Cytoscape对PPI网络进行了可视化,结果见图2;并且通过5种拓扑分析算法鉴定了枢纽基因,其中每种方法选择了前10个基因(表3);最终鉴定出4个枢纽基因,即PRKDC、RBPJ、VCP、MYH11,见图3。

表3 在cytoHubba中由不同拓扑算法识别的前10个枢纽基因

图3 5种方法识别的前10个枢纽基因的韦恩图
2.7 VCP基因突变编码蛋白三级结构预测根据基因-表型评分,确定VCP是最突出的枢纽基因,考虑作为肾小管间质性肾病的候选致病基因。VCP基因的14号外显子的c.1712_1713delT导致氨基酸发生移码,提前出现终止密码子,导致大部分的氨基酸序列丢失。分别构建野生型和c.1712_1713delT突变型蛋白三维结构,预测结果显示:与野生型相比,突变型蛋白大部分氨基酸序列丢失,造成蛋白质空间结构和功能改变,见图4。

图4 野生型(A)和c.1712_1713delT突变型(B)蛋白三维结构
3 讨论
慢性肾脏病影响全球约10% 的成年人,在高危人群患病率已高达50%[9]。大约23%的透析患者的近亲患有终末期肾病,提示慢性肾脏病的遗传原因可能还未被充分认识。近期的研究[10]表明,慢性肾脏病的遗传率在 25%~44%。肾小管间质纤维化是慢性肾脏病的标志,并与疾病进展高度相关。因此,探究单基因与肾小管间质纤维化之间是否相关具有极大临床意义。
本研究针对可能导致肾小管间质性肾病的遗传学机制进行初步探索。在24例肾小管间质性肾病中,发现10%患者共有61个罕见变异,其中35个与肾小管间质性肾病表型存在关联,应用生物信息学工具将上述基因进行通路富集分析,发现其较集中地富集于离子跨膜转运的调节、蛋白质转运的调控等通路上,故该类型通路上的基因发生突变,可能造成肾小管间质性肾病表型。我们进一步构建了一个PPI网络,并根据5种不同的排序方法确定了4个枢纽基因。根据基因-表型评分,发现VCP是最突出的枢纽基因,考虑将其作为肾小管间质性肾病的候选致病基因。
VCP基因c.1712_1713delT在正常人群数据库频率较低。软件预测结果表明野生型与突变型蛋白之间的差异对蛋白质结构和功能产生影响。VCP基因编码含缬氨酸的蛋白质,其主要功能是维持内质网的完整性,并已被证明与内质网相关蛋白质降解途径相关,同时还参与多种细胞过程,包括泛素依赖性蛋白质降解、应激反应和程序性细胞凋亡[11]。内质网是参与蛋白质合成和输出的主要真核细胞器。内质网中不需要或错误折叠的蛋白质被输出到细胞质中,并通过内质网相关蛋白质降解途径被蛋白酶体降解。内质网相关蛋白质降解途径被干扰会导致内质网应激,造成错误折叠蛋白质积累[12]。如果细胞不能从内质网应激中恢复过来,就会发生细胞凋亡[13]。此外,先前的研究[14]表明,内质网应激可能会诱导细胞分化的途径和形态变化。既往研究[15]已建立与内质网应激相关的遗传性肾纤维化小鼠模型,以UMOD基因突变小鼠为实验对象,用于研究肾纤维化、终末期肾病的单基因病因。
综上所述,对于原因不明的肾小管间质性肾病患者,我们应首先考虑是否为遗传性,临床上需注意询问家族病史,并尽早行基因筛查,以避免应用皮质类固醇和免疫抑制剂的治疗。本文对基因-表型相关性的研究,完善了对肾小管间质性肾病遗传学病因的认识,并提出VCP基因可能与遗传相关肾小管间质性肾病有关,为遗传性肾小管间质性肾病的诊断、鉴别诊断及治疗提供新的思路。
猜你喜欢
解放军医学杂志(2021年12期)2022-01-18
现代临床医学(2021年1期)2021-01-26
昆明医科大学学报(2020年12期)2021-01-26
益寿宝典(2018年22期)2018-01-26
安徽医科大学学报(2016年12期)2017-01-15
中国卫生标准管理(2015年7期)2016-01-15
中外医疗(2015年11期)2016-01-04
医学研究杂志(2015年8期)2015-06-22
医学研究杂志(2015年8期)2015-06-22
医学研究杂志(2015年12期)2015-06-10
