
全基因组测序确诊氨基甲酰磷酸合成酶缺乏症Ⅰ型1例
2023-06-24 07:58马鹏飞许愿愿赵晓玲童文佳龙云金丹群吴成陆怡
肝脏 2023年5期
马鹏飞 许愿愿 赵晓玲 童文佳 龙云 金丹群 吴成 陆怡
作者单位:231500 合肥 安徽省儿童医院消化科(马鹏飞,龙云,吴成);安徽省儿童医院重症医学科(许愿愿,赵晓玲,童文佳,金丹群);复旦大学附属儿科医院肝病中心(陆怡)
患儿,男,1个月20天,因“呕吐3天,昏睡、呻吟半天伴反复抽搐发作”入院,患儿系G2P2,足月顺产,否认出生时有窒息抢救史,出生体重3 400 g,父母体健,否认近亲结婚,否认家族遗传病、传染病及肝胆疾病史。患儿哥哥体健。入院查体:昏睡状态,精神、反应差,面色稍苍白,无特殊面容,全身皮肤无黄染、淤点淤斑、出血点及花纹征,呼吸深快,三凹征(-),心肺听诊阴性,腹膨隆、软,腹壁未见静脉曲张,肝脾肋下未及,肠鸣音正常,四肢肌张力增高,四肢阵发性强直阵挛发作。双下肢无浮肿,双侧足背动脉搏动有力。急诊血气分析:PH 7.51,PCO222.3 mmHg,PO285mmHg,cHCO215.5 mmol/L,BE -7.23 mmol/L,AG 13.1 mmol/L;予以禁食水(不禁药)、吸氧、心电监护、止惊、复合氨基酸营养、补液等对症处理,同时口服维生素B6、维生素B12及左卡尼丁。肝功能:TB 44.6 μmol/L(2-21 mmol/L),DB 14.7 μmol/L(0-8 mmol/L),ALT 28.7 IU/L(5-60 U/L),AST 25.1 IU/L(5-60 U/L),肾功能、电解质、血糖、CRP及免疫球蛋白正常;凝血五项、Torch、嗜肝病毒、梅毒、HIV、丙酮酸、同型半胱氨酸正常,胸部X线片及头颅CT正常,急诊行脑脊液常规及生化检查,排除了中枢神经系统感染;测血氨134.8 μmol/L,停止氨类摄入,“精氨酸”静滴补充氨基酸,当晚出现呼吸困难,转PICU机械通气,同时复测血氨231.8 μmol/L,予以限制氨摄入及促进肠道氨排出,未见好转,血氨升高至466.0 μmol/L,给予持续性肾脏替代治疗(CRRT)27 h后,血氨下降至41.1 μmol/L,第三日鼻饲开奶后出现病情加重,持续昏迷,复测血氨151 μmol/L,凝血五项:PT 32.4 s(10~14 s),INR2.88(0.85~1.45),APTT>180 s(23~40 s),TT>180 s(14~21 s),FBG2.45 mg/L(0~0.5 mg/L),再行CRRT。第四日血串联质谱提示:瓜氨酸 5.03(9.63~64.17),丙氨酸175.38(102.86~450),精氨酸12.77(1.50~55.00),尿串联质谱提示:乳清酸0;乳酸-2 233.38(0~4.7),3-羟基丁酸 24.73(0~3.7),丙酮酸-OX 108.47(0~24.0)提示酮症,考虑为CPS1D,但不能排除N-乙酰谷氨酸合成酶缺乏症,与家长沟通,征求家长同意后行基因检测;第五日,患儿出现对光反射消失,行头颅CT提示脑水肿、脑疝及蛛网膜下腔出血(图1),同时血常规:WBC 17.7×109,HB 53 g/L,血小板下降至30×109,CRP330 mg/L,使用美罗培南联合利奈唑胺抗感染治疗及对症处理,复测血氨上升达238.5 μmol/L,再次行CRRT。第六日,肝功能:TB 27.4 mmol/L,DB 10.9 mmol/L,ALT 97 U/L,AST 66 U/L,胆碱酯酶 3198 U/L,电解质:Na+199 mmol/L,Cl-171 mmol/L,K+5.77 mmol/L,持续葡萄糖水维持中,血糖1.5 mmol/L,于第七日家长放弃治疗,出院后死亡。

图1 A. 入院第一天头颅CT:脑实质内未见异常密度影。B. 入院第5天头颅CT:双侧大脑半球脑实质密度减低,灰白质分界不清,脑沟裂变浅,环池、脚前池、四脑室明显变窄、消失,大脑纵裂池、小脑上池密度明显增高,双侧侧脑室受压变窄消失,中线结构无明显移位。小脑扁桃体疝入枕骨大孔。蛛网膜下腔出血
基因检测结果:CPS1基因复合杂合突变:exon11:c.1145C>T:p.P382L ,c.1360-189T>C,经Sanger测序验证分别来自父亲和母亲(图2)。本文中c.1145C> T:p.P382L位点为已报道的致病突变,c.1360-189T>C位点经SpliceAI和varSEAK在线分析平台对变异的剪接功能生物学危害性预测均为阴性,但根据美国医学遗传学与基因组学学会 (The American College of Medical Genetics and Genomics,ACMG)指南,该变异可评级为可能致病性变异: PM2(正常人群变异数据库未见报道)+PM3(反式位置有致病变异)+PP1(突变与疾病在家系中共分离)+PP4(临床表型高度符合)。
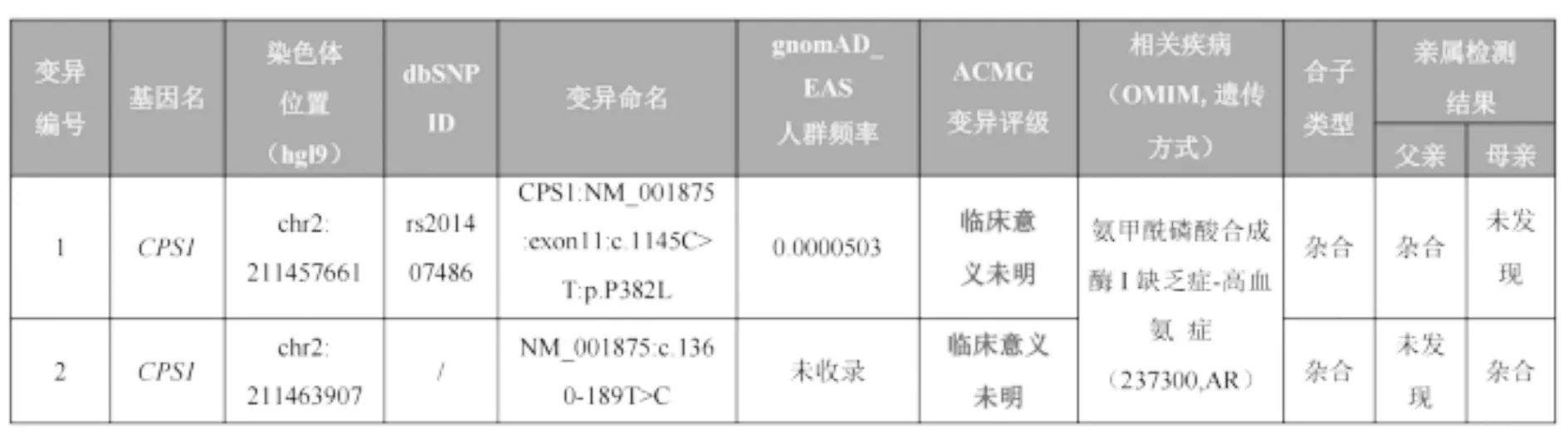
图2 患儿家系的全基因组测序结果
讨论氨基甲酰磷酸合成酶缺乏症Ⅰ型(CPS1D;OMIM #237300)是一种常染色体隐性遗传病,是因尿素循环中的第一个限速酶:氨基甲酰磷酸合成酶-1(CPS1)缺乏或者活性降低,导致氨基酸代谢异常而表现的一系列临床症状[6, 18]。该病发病率低,可起病于任何年龄,大多数病例起病于新生儿时期,肝脏代谢异常,以高氨血症的神经症状为主要临床表型,重症者病情进展快,病死率高,难以马上确诊,故临床确诊病例罕见。
本研究分别以“Carbamoyl Phosphate Synthetase 1 Deficiency”和“氨基甲酰磷酸合成酶缺乏症Ⅰ型”在PubMed以及万方数据库和中国知网中检索,时间限制在2022年3月1日之前,大陆地区已报道的文章共16篇,英文发表的文章有8篇[1-8],中文发表的文章有8篇[9-16],与21例患儿相关(排除重复报道3例),新生儿期发病的病例15例,最早发病时间为出生后1天;迟发型病例6例,最迟发病时间为11岁9个月,临床表型无特异性,血氨733.3±794.4 μmol/L(81~1404 μmol/L)。基因突变位点共累及32个,其中外显子31个,内含子仅1个,为c.622-3C>G;错义突变24个,无义突变5个,小片段缺失1个,移框突变1个,剪接突变1个。包括本文中的突变位点,c.1145C>T的错义突变出现频率最高,共3次;相同位点的复合杂合突变仅一对,即: Exon19: c.2339G>A; Exon29: c.3520C>T,由此验证了CPS1基因具有的“私有化”特征[4, 17]。经OMIN、HGMD网站查询,本文中的内含子c.1360-189T>C的错义突变尚未报道。
CPS1作为尿素循环的第一个限速反应,催化氨进入循环[12],该酶由CPS1(MIM*608307)基因编码。CPS1基因位于染色体2q35上,跨度>120kb,包含超过38个编码外显子的4500个编码核苷酸[19],其变异可使CPS1的功能降低或缺失,导致高氨血症。随着血浆氨水平的升高,纳差、呕吐、呻吟、惊厥发作、呼吸窘迫和反常呼吸等呼吸不适的发生率增加,这些可能是高氨血症引起的水肿刺激脑干呼吸中枢的结果[20]。
鉴于许多患者在确诊前死亡,CPS1D的确切发病率尚不清楚,据估计,日本的发病率约1/80万,芬兰的发病率约1/53.9万[6]。在尿素循环障碍(UCDs)中CPS1D的病死率最高,其次是OTCD和ASA[20]。在新生儿期发病的CPS1D和OTCD患者中,60%患者在新生儿期能存活,但其中约19%的存活儿在1年内因再次或反复病情发作而死亡。对于CPS1D患者,在第一年内病情反复可能高达8次。即使患者在第一年存活下来,约47%~67%的患者存在发育迟缓,只有15%~20%的患者发育正常[21]。发育迟缓的程度与高氨血症的峰值浓度和持续时间有关[18],所以早期诊断及早期治疗对患者至关重要。
众所周知,同一疾病不同基因突变的患者可能表现出不同的表型。从表1中可以看出,CPS1D的临床表型虽无特异性,但可因病情的轻重缓急而表现出不同程度的神经系统症状。特别对于新生儿期发病的患者来讲,发病早,病情进展快,病死率高,如果临床医生对该病没有充足的认识,则很难诊断该病。CPS1D的诊断基于血氨升高、瓜氨酸降低、尿液中的乳清酸正常或较低[10]。然而,由于UCDs中的N-乙酰谷氨酸合酶缺乏症(NAGSD)有着与CPS1D相类似的中间代谢物,所以该技术无法将CPS1D与NAGSD区分开来[5],且这些中间代谢产物易受感染、饮食及其他代谢异常等影响[21],故明确诊断需要对肝脏或小肠组织进行酶学分析,或者进行分子遗传学研究[22]。本例患儿有严重高氨血症伴血、尿串联质谱典型表现,基因分析排除了NAGS基因缺陷,故CPS1D诊断成立。
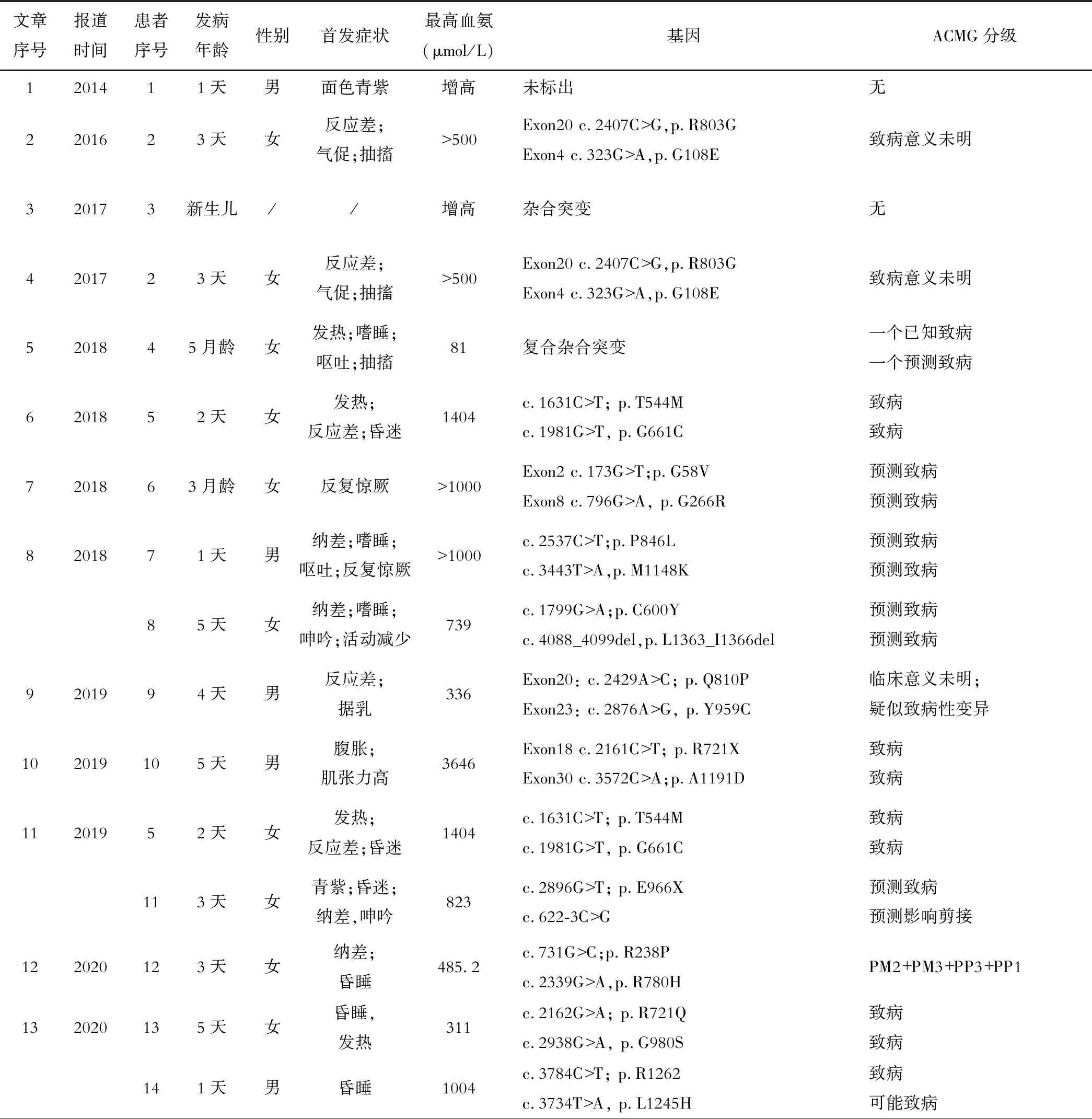
表1 大陆地区已发表的CPS1D患儿首发表现及基因突变位点
该患儿二代测序(NGS)仅测出CPS1外显子c.1145C>T,来源于父亲,为已报道致病突变,随后再次行WGS检测,得出第二个突变位点,内含子c.1360-189T>C,来源于母亲,其哥哥WGS检测仅得出CPS1外显子c.1145C>T的杂合子。经OMIN、HGMD网站查询,内含子c.1360-189T>C突变位点未曾报道。根据表1,大陆地区患者中CPS1的突变位点几乎多位于外显子,内含子突变仅有1例,且深度较浅,本文中的突变位点在内含子的较深处。瑞士的Jasmine Isler报道过12个CPS1的内含子深部突变,其中最深位置c.3559-745A>G,他对高度怀疑CPS1D而NGS阴性或者杂合子的患者增加RNA测序和(或)酶学分析,确诊了多例怀疑该病的病例,还解释了内含子的突变可对剪接功能有影响[22]。本文中的患儿临床表型符合CPS1D,ACMG评估为可能致病,再结合CPS1基因具有较高的“保守性”,故CPS1的变异极可能导致致病[17, 22],但遗憾的是,家长拒绝行RNA测序及肝脏或小肠组织的酶学分析。表1中所涉及的21位患者中已标出基因突变者19例,累及突变位点32个,且仅有1对相同的复合杂合突变,大多数突变并不重复,这表明了CPS1基因突变的“私有化”特征,进一步使CPS1D的诊断复杂化[17]。因此临床上对于高度怀疑CPS1D的患儿需行NGS或WGS检测,同时需关注内含子深处的突变,必要时可行RNA测序及酶学分析验证诊断。
CPS1D是一种罕见的临床表型无特异性的疾病,早期极易误诊为感染性疾病。表1中21例病例在入院时明确有感染的7例,另外有7例怀疑为感染性疾病。本文患儿在入院时纳差、呼吸节律改变、反复惊厥发作,不能排除中枢神经系统感染,所以使用“苯巴比妥”镇静和“复合氨基酸”营养支持治疗,但这两种药物都会加重血氨的升高。除此之外,丙戊酸、卡马西平、托吡酯及左旋门冬酰胺等亦会加重血氨升高,所以对于高氨血症患者应避免使用[4]。CPS1D的病程可呈间歇性发作,除上述的特殊药物可诱发该病发生外,应激状态、病毒感染或高蛋白饮食亦会诱导病情发作及导致病情恶化[8]。本文中的患儿在第一次CRRT后,血氨已降至正常,但开奶后患儿再次出现血氨升高。GRIFFIN 等人曾在模拟实验中证明CPS1活性低于50%正常值时,在推荐蛋白质摄入量下血氨水平增加1倍以上,高蛋白饮食下血氨水平增加3倍以上[23]。脑氨水平的增加会阻碍星形胶质细胞对钾的吸收,导致神经元吸收钾增加,从而损害皮质的抑制性神经传递,导致癫痫发作[23]。
患者昏迷持续时间的延长和血氨水平的增加被认为是导致发病率和病死率增加的原因[21]。在死亡的患者中,高血氨状态持续时间的中位数为4天半,而存活者中高血氨状态持续时间的中位数为3天[20]。该患儿在持续3天多的高氨血症后出现了急性肝功能衰竭[24],随着病情的进展,在“肝衰”的晚期并发了多脏器功能衰竭,如:重症感染、骨髓造血功能抑制、脑水肿、脑疝、蛛网膜下腔出血及严重的电解质紊乱,所以及时降低血氨被认为是预防不良后果的关键。CPS1D的治疗现无专家共识和指南参考,主要依据尿素循环障碍的治疗方案[21]。目前,有几种新的治疗方法正在研发中,如:N-氨甲酰-L-谷氨酸(NCG)的利用;药物伴侣增加错误折叠突变蛋白的稳定性;基因治疗及黄酮类化合物等研制[25]。
总之,本文拓展了CPS1基因突变的基因谱。鉴于CPS1D的非典型症状、突发性、快速进展和低发病率,许多CPS1D患者在确诊前已死亡。本文总结了大陆地区报道的儿童CPS1相关UCDs的临床和生化特征,提醒临床医生在遇到喂养后出现纳差、癫痫发作和意识障碍等神经系统表现的患儿,需行血氨测定。一旦确诊高氨血症,应合理饮食及避免使用加重高氨血症的相关药物,尽早行血尿串联质谱分析和NGS,基因检测。不能忽略内含子的突变,即使检测结果为阴性,但临床高度怀疑CPS1的患者可行RNA测序或酶学检测确诊;早期诊断、早期治疗,有助于降低病死率和改善预后。由于本文CPS1缺陷例数少,可能存在观察偏移。
利益冲突声明:所有作者均声明不存在利益冲突。
猜你喜欢
内蒙古师范大学学报(自然科学汉文版)(2021年3期)2021-06-01
首都食品与医药(2021年5期)2021-03-22
生物工程学报(2019年6期)2019-07-10
生物学通报(2019年1期)2019-02-15
生物学通报(2018年12期)2018-10-10
转化医学电子杂志(2018年11期)2018-01-16
现代园艺(2017年21期)2018-01-03
中国现代药物应用(2016年10期)2016-03-06
中国康复理论与实践(2015年10期)2015-12-24
医学研究杂志(2015年5期)2015-06-10
