
线粒体功能障碍与血管平滑肌细胞表型转化的研究进展
2023-05-10 06:07瞿珊珊黄蓉蓉闫军宇李玉兰
心血管病学进展 2023年4期
瞿珊珊 黄蓉蓉 闫军宇 李玉兰,3
(1.兰州大学第一医院生殖医学中心,甘肃 兰州 730000; 2.兰州大学第一临床医学院,甘肃 兰州 730000; 3.兰州大学第一医院麻醉科,甘肃 兰州 730000)
心血管疾病的患病率逐年上升,在人类疾病死亡率中居首位。20世纪90年代,Takaichi等[1]发现血管平滑肌细胞(vascular smooth muscle cells,VSMCs)具有两种表型——收缩型和合成型(或分泌型),且在一定条件下可互相转化。近年来研究[2-8]发现,VSMCs表型转化在心血管疾病的发生和发展中起关键作用,而线粒体功能障碍、钙稳态失衡可能是VSMCs表型转化的重要因素[9-15]。现综述钙稳态、线粒体功能与VSMCs表型转化之间的关系,并探讨其促成心血管疾病的可能机制。
1 VSMCs表型转化与心血管疾病
VSMCs是血管中膜的主要成分。生理情况下,VSMCs分化成熟为稳定的收缩型,维持血管正常的舒缩功能。当受到病理因素刺激时,VSMCs由收缩型去分化为合成型,获得较强分泌功能,合成分泌细胞外基质,细胞增殖和迁移能力增强,导致内膜增生和血管重构[2-3]。VSMCs不同表型具有相关标志性蛋白,收缩型标志性蛋白有α-平滑肌肌动蛋白(alpha-smooth muscle actin,α-SMA)、平滑肌蛋白22α(smooth muscle 22 alpha,SM22α)、平滑肌肌球蛋白重链和钙调蛋白等;合成型标志性蛋白有骨桥蛋白(osteopontin,OPN)、骨形态发生蛋白质2和骨钙素等。合成和分泌能力可通过检测基质金属蛋白酶2、基质金属蛋白酶9等活性物质进行鉴定[4]。
VSMCs表型转化引起的功能障碍是心血管疾病的病理基础,如动脉粥样硬化、动脉高压、血管钙化、血栓形成和动脉瘤形成等。在动脉粥样硬化的病理改变中VSMCs表现出较强的分化增殖能力,细胞形态向软骨细胞样、成骨细胞样、泡沫细胞样、巨噬细胞样转化,收缩表型蛋白表达降低的同时合成表型蛋白明显升高,驱动血管钙化形成。因此认为VSMCs表型转化是启动动脉粥样硬化病变的重要因素[5]。肺动脉高压(pulmonary hypertension,PH)种类繁多,发病机制复杂,但各类PH具有相同的病理标志即血管重塑,表现为肺动脉血管平滑肌细胞(pulmonary arterial vascular smooth muscle cell,PASMC)增殖,血管内膜增厚,血管肌化明显。在各种病因引起的PH病变血管中,均发现PASMC存在去分化现象,细胞过度增殖、迁移,导致血管病理性重塑。在高血压病变血管中VSMCs增生明显,中膜增厚,血管僵硬,导致血管阻力增加,收缩标志蛋白α-SMA、SM22α表达下调,合成标志蛋白OPN表达增加,表现出明显的转化现象[6-8]。
2 线粒体功能与VSMCs表型转化
线粒体是细胞内重要的细胞器之一,具有为细胞供能、参与信号转导、维持细胞内钙稳态、产生活性氧(reactive oxygen species,ROS)、调节细胞凋亡等功能。它具有高度动态性,可通过融合及分裂的动态平衡,调节能量代谢,保证细胞正常的生理功能。近期研究VSMCs表型转化时发现,线粒体形态及功能发生改变,其动力学及能量学的稳定在维持血管平滑肌表型及功能方面具有重要作用。
2.1 线粒体动力学
线粒体是一个不断进行融合分裂的动态管状网络,其融合过程具有混合并重新分配蛋白质、代谢产物和线粒体DNA等功能;分裂过程则是将线粒体网络中功能失调或受损的部分分离出来,通过自噬进行降解,从而修复受损的线粒体[9]。
在VSMCs表型转化过程中,电镜观察到线粒体形态变化显著[10],收缩表型VSMCs中线粒体呈现蠕虫形丝状,而合成表型中则表现为球形的碎片化状态。线粒体的重塑过程受相关蛋白调控,如促进融合的Mitofusin1/2(Mfn1/2)、视神经萎缩蛋白等和促进分裂的动力相关蛋白1(dynamin-related protein 1,Drp1)、分裂蛋白1等。线粒体融合后,可恢复膜电位,增加氧消耗率和三磷酸腺苷(adenosine triphosphate,ATP)的产生[11-12]。而线粒体分裂,膜去极化,电位降低,能量代谢障碍,诱导线粒体自噬。
Mfn2介导线粒体融合,其正常表达是维持VSMCs收缩表型的重要因素。当Mfn2表达减少时,VSMCs发生表型转化,增殖能力增强。在缺氧性PH的大鼠模型中Mfn2转录及蛋白表达显著降低,VSMCs增生,血管壁增厚[13]。血小板源性生长因子BB(platelet-derived growth factor-BB,PDGF-BB)是诱导VSMCs去分化的重要生物因子[14]。Salabei等[10]在研究PDGF-BB调控VSMCs表型转化时观察到Mfn2的蛋白表达减少50%,线粒体分裂活性增强,呈现明显的碎片化,细胞中α-SMA和钙调蛋白表达减少。Torres等[15]在PDGF-BB诱导大鼠主动脉VSMCs表型转化时,使用胰高血糖素样肽-1激活蛋白激酶A活性,上调Mfn2的表达,促进线粒体融合,抑制细胞迁移和增殖。牛艳华[16]发现脂肪酸合酶抑制剂可增加Mfn2的表达,减轻线粒体相关的细胞凋亡,改善血管重塑。
相反,线粒体分裂增强可诱导VSMCs向合成型转化。在慢性血栓栓塞性PH的VSMCs中,Drp1磷酸化显著增加,线粒体体积减小,数量增加,OPN上调,SM22α显著下调[17]。Marsboom等[18]通过激活缺氧诱导因子-1α,诱导线粒体Drp1总量增加、磷酸化增强,VSMCs获得较强的增殖能力,导致内膜增厚。生物因子血管紧张素Ⅱ(angiotensin Ⅱ,AngⅡ)也可通过增加Drp1、降低Mfn2的表达,使VSMCs表型转化[19]。但Deng等[20]在AngⅡ诱导小鼠原代主动脉VSMCs表型转化过程中发现,Drp1的mRNA及蛋白表达没有增加,而是通过AngⅡ增强Drp1磷酸化,激活Drp1发挥生物效应。无论通过何种机制,在使用线粒体分裂抑制剂Mdivi-1后,均可观察到线粒体面积和长宽比明显增加,同时增殖标志物减少,AngⅡ诱导的表型转化被明显抑制,逆转VSMCs去分化现象,恢复收缩表型[18-20]。
VSMCs表型转化伴有线粒体动力学的改变,线粒体融合减弱,分裂增强。改善线粒体功能,可逆转去分化以维持VSMCs收缩表型,改善血管功能,证实线粒体动力学在VSMCs表型转化中具有重要的调控作用。
2.2 线粒体能量学
线粒体是产生能量的重要细胞器,其内膜包含电子传递链和线粒体ATP酶,参与三羧酸循环的氧化磷酸化和β氧化,为机体供能[11]。线粒体损伤时,氧化磷酸化受到抑制,ATP产生减少,线粒体功能障碍,大量生成ROS,线粒体DNA(mitochondrial DNA,mtDNA)损伤,钙稳态紊乱,导致一系列的心血管疾病[21]。
近期研究发现,VSMCs表型转化时伴有明显的能量代谢障碍。在小鼠主动脉合成型(去分化型)VSMCs的组织模型中,观察到参与氧化磷酸化的复合物Ⅰ、Ⅲ、Ⅴ的表达明显降低,复合物Ⅱ、Ⅳ也表现为下降趋势,有氧糖酵解与氧化磷酸化耦合减弱,能量代谢障碍[22]。Yu等[23]也发现在动脉粥样硬化斑块的VSMCs中复合物Ⅰ表达减少,线粒体呼吸功能受损,ATP产生减少,这可能与mtDNA损伤有关。mtDNA是控制呼吸链蛋白合成的重要分子,其损伤使编码的线粒体ATP酶的表达降低,氧化磷酸化功能受损,ATP合成显著减少,直接影响能量代谢[24-25]。
线粒体能量代谢障碍也可诱导VSMCs表型转化。软骨寡聚基质蛋白与抑制素蛋白2相互作用维持VSMCs的收缩表型。当线粒体内的软骨寡聚基质蛋白缺乏时,线粒体膜电位降低,mtDNA损伤,出现明显的氧化磷酸化障碍,VSMCs向合成型转化[26];抑制素蛋白2缺乏时,抑制复合物Ⅰ、Ⅱ、Ⅳ的活性,损害呼吸超复合物形成,导致相同的结果[27]。寡霉素抑制ATP合酶(又称复合体Ⅴ)时,也观察到VSMCs去分化的现象。在PDGF-BB导致的合成表型VSMCs中,Jia等[26]观察到去分化程度与线粒体呼吸减少呈正相关;相应的在转化生长因子-β诱导的收缩表型中,线粒体呼吸明显增加。
正常收缩型细胞线粒体主要利用葡萄糖代谢供能,VSMCs去分化时,线粒体氧化磷酸化减弱,能量代谢障碍。为保证细胞能量代谢,脂肪酸逐渐成为主要供能物质。Salabei等[10]研究认为脂肪酸氧化是细胞分裂、迁移及合成和分泌细胞外基质的必需环节,VSMCs去分化获得增殖能力是由转录和生物能量过程耦合驱动的。因此,改善线粒体呼吸功能、恢复供能,可成为恢复细胞表型的治疗手段。线粒体能量代谢与其形态相关,高能量需求的组织(如骨骼肌、心肌细胞等)通常具有融合的相互连接的线粒体,而能量需求较低的组织有更小、碎片化的线粒体[28]。胰高血糖素样肽-1通过上调Mfn2的表达快速诱导线粒体融合,线粒体膜电位、氧消耗率和ATP增加,线粒体活性增强,抑制细胞的增殖和转移。因此认为线粒体的融合是对能量代谢失调的一种保护性反应[17]。
3 钙离子与线粒体功能
线粒体是细胞内重要的钙离子缓冲和储存器,细胞内钙平衡是维持VSMCs正常生理功能的保障。在PH患者的VSMCs中观察到线粒体断裂、线粒体内钙离子浓度病理性降低,能量代谢受抑制;细胞质中钙离子浓度升高,驱动细胞增殖、血管收缩[29]。人们通过电镜观察发现,在内质网膜上的钙离子通道附近通常有线粒体聚集,参与介导钙离子重新分布,维持细胞质内的钙离子稳态,保证正常的细胞功能[30],如图1。
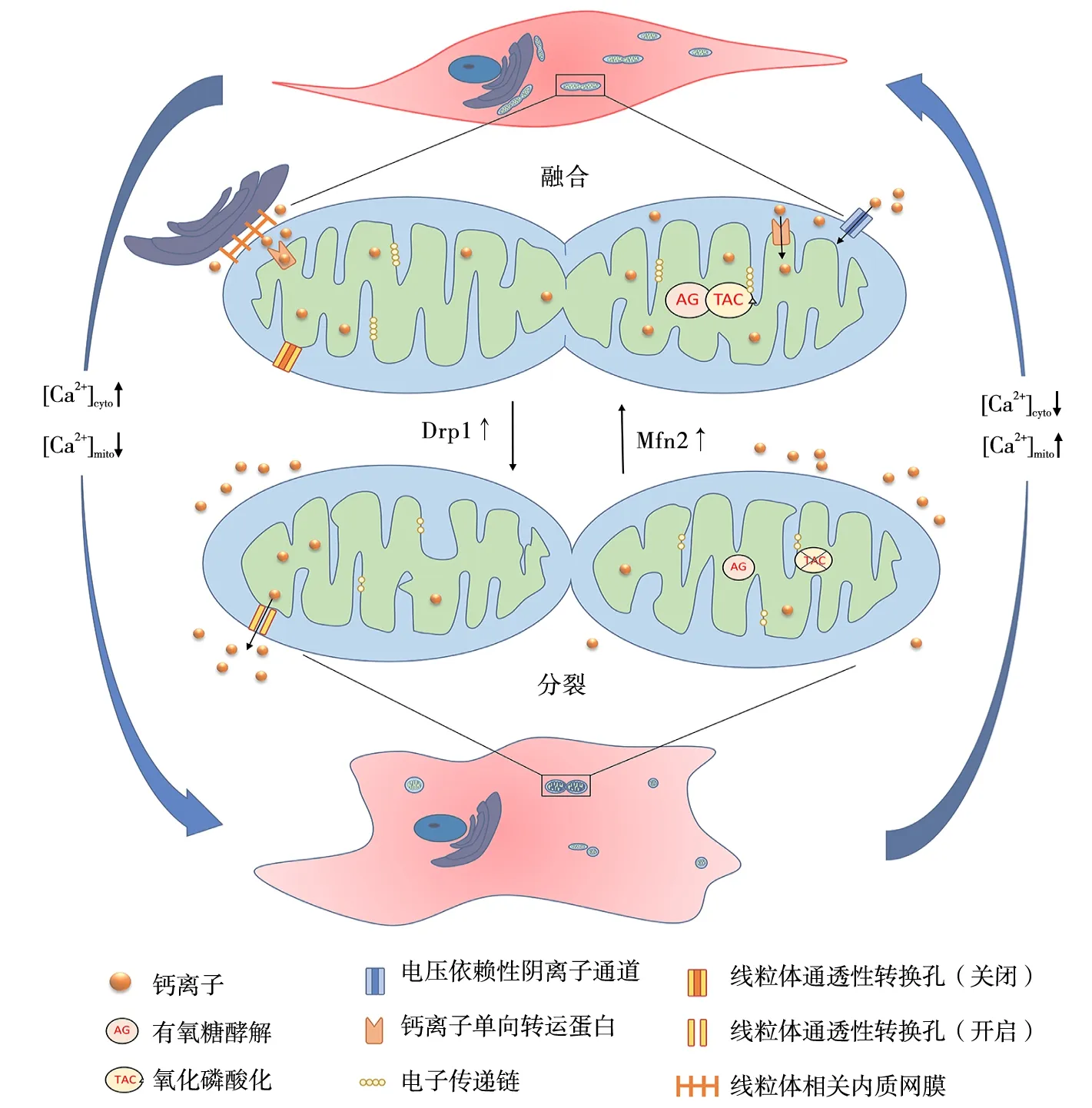
注:[Ca2+]cyto,细胞质钙离子浓度;[Ca2+]mito,线粒体钙离子浓度。
线粒体对钙离子的摄取有赖于内质网与线粒体的偶联区域,即线粒体相关内质网膜(mitochondria-associated endoplasmic reticulum membranes,MAMs)[31]。MAMs主要功能包括产生高度定位和集中的钙离子微区,促进钙离子通过电压依赖性阴离子通道穿过线粒体外膜到达线粒体膜间腔隙,激活线粒体钙离子单向转运蛋白(mitochondrial calcium uniporter,MCU)进入线粒体基质内[32]。线粒体能量代谢对细胞内钙离子浓度具有依赖性。钙离子浓度适度升高激活MCU,钙离子进入线粒体,促进能量合成。线粒体内的钙离子浓度决定ATP的合成速率[33]。Bravo等[31]实验观察到在内质网应激早期,MAMs数量增加,确保内质网与线粒体间的钙联系。内质网释放钙离子激活MCU,提高线粒体钙离子摄取率,降低细胞质内钙离子浓度,同时确保ATP的产生。在VSMCs中这种作用有利于维持VSMCs的收缩表型[34]。
研究[32]发现,Mfn2在MAMs中高度表达,是重要的调节蛋白,其表达参与调节线粒体的钙摄取。Mfn2表达增加,线粒体融合,提高对钙离子摄取能力及速率[35]。Mfn2表达减少,内质网与线粒体之间的距离增加,线粒体钙离子摄取率降低[36],ATP生成障碍,细胞质内钙离子浓度升高,诱导VSMCs表型转化。Hong等[29]在正常的PASMC中,通过基因调控抑制MCU活性,细胞质内钙离子浓度升高,Mfn2表达降低,线粒体碎片化,细胞增殖、迁移能力增强。同时,线粒体内钙离子浓度降低,抑制丙酮酸脱氢酶活性,氧化代谢减弱,细胞功能依赖有氧糖酵解,出现类似于癌细胞的Warburg现象,能量代谢障碍,表现出合成型VSMCs的特征。抑制Drp1及其磷酸化,分裂过程减弱,也可使线粒体获得更高的钙摄取和保留能力[35]。
除此之外,ROS和炎症因子等致病因子导致线粒体钙超载,每毫克线粒体中钙离子含量>500 nmol时,线粒体氧化磷酸化受到不可逆抑制,ATP产生明显减少,可能与线粒体内膜嵴功能障碍[37]和复合物Ⅰ功能抑制有关[38]。同时,钙超载导致线粒体通透性转换口打开,膜电位降低,线粒体肿胀,内膜断裂嵴重塑,mtDNA损伤,线粒体碎片化,自噬增强,能量代谢障碍[39]。线粒体通透性转换口的持续开放导致线粒体钙离子和ROS流入细胞质内,细胞质内钙离子和ROS浓度升高诱导VSMCs表型发生转化。线粒体膜电位降低和呼吸链功能缺陷有关的钙摄取能力下降,也是诱导VSMCs表型转化的机制之一[32]。
钙离子是维持线粒体动力学及能量学的重要桥梁,VSMCs表型转化过程中线粒体功能障碍表现为:动力学失衡,形态呈碎片化,钙离子摄取能力降低,细胞内钙离子浓度升高,线粒体内钙离子浓度降低,能量代谢障碍。病理因素刺激下,线粒体钙超载抑制氧化磷酸化,并导致线粒体肿胀、膜断裂等,诱导VSMCs向合成型转化。因此,保证钙稳态是维持线粒体功能的重要环节。
4 结论与展望
综上所述,VSMCs表型转化是多数心血管疾病发病的基础改变,线粒体功能状态是决定VSMCs表型的重要因素,线粒体融合强于分裂,利于氧化磷酸化及ATP的产生,VSMCs表达为收缩型。钙离子是影响线粒体功能的核心因素之一,钙离子浓度与线粒体的融合分裂过程及能量代谢相互作用,共同影响VSMCs的表型。因此如何维持细胞内钙稳态,改善线粒体功能,使VSMCs从病理状态下的合成型恢复为收缩型,可能成为阻断心血管病变发展的治疗策略。
猜你喜欢
青岛大学学报(医学版)(2022年3期)2022-12-27
天津医科大学学报(2019年6期)2019-08-13
中成药(2018年5期)2018-06-06
现代园艺(2017年21期)2018-01-03
分析化学(2017年12期)2017-12-25
中国药理学通报(2017年6期)2017-06-22
中国康复理论与实践(2015年10期)2015-12-24
安徽医科大学学报(2015年9期)2015-12-16
山西医科大学学报(2015年6期)2015-12-16
医学研究杂志(2015年5期)2015-06-10
