
基于TOPSIS法的化胃舒颗粒水提取工艺研究
2022-12-21 08:18王德成邹桂欣王光函
亚太传统医药 2022年8期
王德成,姜 鸿,赵 玥,邹桂欣,王光函
(辽宁省中医药研究院,辽宁 沈阳 110034)
化胃舒颗粒为我院临床常用制剂,由黄芪、麦冬、红参、茯苓、甘草、法半夏、乌梅等十三味中药组成,具有益气健脾、养阴和胃、降逆止呕之功效。现代药理学表明,黄芪、麦冬、红参、茯苓、甘草、法半夏所含皂苷类及多糖类化合物分别具有抗肿瘤[1-6]、增强机体免疫力[7-8]、拮抗化疗药物副作用[9]、修复胃肠道黏膜等活性,乌梅所含有机酸具有促进胃酸分泌、修复胃黏膜损伤、调整脾胃功能、调节胃肠蠕动功能等[10-12]作用。上述化学成分均与本品功能主治相关,起到治疗或者减毒增效作用,且具有一定水溶性。因此拟采用正交实验法,根据现阶段国内药品生产企业水提取设备现状,测定提取液中黄芪甲苷、总多糖、总皂苷、总有机酸的含量,结合逼近理想解排序法(TOPSIS)评价不同工艺条件的优劣。TOPSIS是根据评价对象与理想化目标的接近程度进行顺序优选的一种多指标决策分析方法,其将多维问题转化为一维问题,降低了分析过程中不同类型指标对决策的干扰,可以提高多目标决策分析的科学性和准确性。
本研究将TOPSIS法与正交试验相结合,对化胃舒颗粒水提取的工艺参数进行优化,以期有效提高正交试验结果的科学性。
1 材料与仪器
1.1 试药
黄芪甲苷(批号:110781-200904),D-无水葡萄糖(批号:110833-200904)人参皂苷Rg1(批号:110703-201027)购于中国食品药品检定研究院。乙腈、甲醇(美国迪马公司)为色谱纯,水为娃哈哈纯净水,其余试剂为分析纯。
1.2 仪器
Agilent11000 高效液相色谱仪(美国安捷伦科技公司);蒸发光散射检测器(Alltech ELSD 2000ES);Agilent8453 紫外可见分光光度计(美国安捷伦科技公司);FA1004 电子分析天平(上海上天精密仪器有限公司)。
2 方法与结果
2.1 黄芪甲苷含量测定方法
2.1.1 色谱条件 色谱柱:Kromasil C185 μm,250 mm×4.6 mm(翡纳米科技有限公司);流动相:乙腈-水(37∶63);流速:1 mL/min;蒸发光散射检测器:漂移管温度:105 ℃;气体流速:3.0 L/min。
2.1.2 黄芪甲苷对照品溶液制备 取黄芪甲苷对照品,精密称取10.2 mg,置25 mL量瓶中,加甲醇适量使溶解并稀释至刻度,摇匀,作为对照品储备溶液(浓度为408 μg/mL)。精密吸取对照品储备溶液2.5 mL,置10 mL容量瓶中,甲醇稀释至刻度,摇匀,作为黄芪甲苷对照品溶液(浓度为102 μg/mL)。
2.1.3 供试品溶液制备 精密吸取水提取液25 mL,加水30 mL,摇匀,水饱和正丁醇提取3次,每次25 mL,合并正丁醇液,用氨试液洗涤2次,每次25 mL,弃去氨试液,正丁醇液用正丁醇饱和水溶液洗涤2次,每次25 mL,取正丁醇液,蒸干,残渣加甲醇使溶解,并定量转移至5 mL量瓶中,甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
2.1.4 测定方法 分别精密吸取黄芪甲苷对照品溶液5 μL、20 μL,供试品溶液20 μL,注入液相色谱仪,测定,用外标两点法对数方程计算黄芪甲苷含量。黄芪甲苷对照品溶液和正交试验水提取液供试品溶液HPLC色谱见图1、图2。

图1 黄芪甲苷对照品HPLC色谱
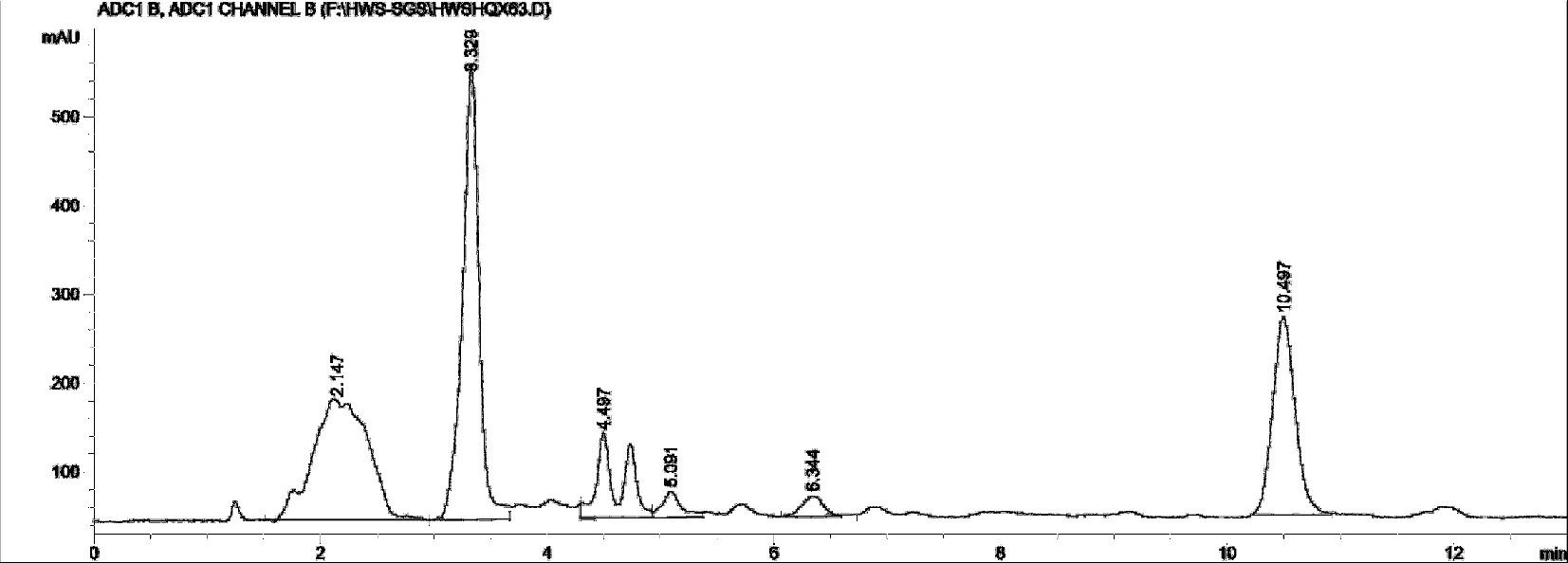
图2 提取液供试品HPLC色谱
2.1.5 供试品精密度试验 精密吸取水提取液25 mL,按“2.1.3供试品溶液制备”项下方法制得供试品溶液,精密吸取供试品溶液20 μL,按“2.1.4测定方法”项下实验,连续测定6次。其结果见表1。

表1 精密度实验结果
2.1.6 重复性试验 精密吸取提取液25 mL,平行6份,分别按“2.1.3供试品溶液制备”项下方法制得供试品溶液,精密吸取供试品溶液20 μL,按“2.1.4测定方法”项下实验。其结果见表2。

表2 重复性实验结果
2.2 总有机酸含量测定方法研究
2.2.1 氢氧化钠滴定液配制 取氢氧化钠适量,加蒸馏水振摇使成饱和溶液,冷却后,置聚乙烯塑料瓶中,静置数日,澄清后备用。取澄清后的氢氧化钠饱和溶液5.6 mL,加新沸过的冷水使成1 000 mL,摇匀,得氢氧化钠滴定液(0.1 mol/L),备用。
标定:取在105 ℃干燥至恒重的基准邻苯二甲酸氢钾约0.6 g,平行3份,精密称定,加新沸过的冷水50 mL,振摇,使其尽量溶解;加酚酞指示液2滴,用氢氧化钠滴定液滴定,滴定至溶液显粉红色。每1 mL氢氧化钠滴定液(0.1 mol/L)相当于20.42 mg的邻苯二甲酸氢钾。计算F值,计算公式如下:
F=W/(20.42 mg/mL×10-3V)(W:邻苯二甲酸的称样量;V:消耗的氢氧化钠的体积)
3次平行试验测得F的平均值为1.004。
2.2.2 硫酸滴定液配制 取浓硫酸1.2 mL,缓缓注入适量蒸馏水中,冷却至室温,加蒸馏水稀释至1 000 mL,摇匀,得硫酸滴定液。标定:取新配制的0.1 mol/L氢氧化钠溶液10 mL,加水50 mL,混匀后,加入甲基橙试液2滴,用0.02 mol/L的硫酸滴定液滴定。平行操作5次,计算消耗硫酸滴定液体积的平均值,标定硫酸滴定液的浓度。
2.2.3 水提取液中总有机酸测定 分别精密吸取正交实验各次实验水提取液15 mL,按照2015版《中华人民共和国药典》四部电位滴定法与永停滴定法(通则0701)实验。将水提取液置烧杯中,加水50 mL,摇匀,精密加入0.1 mol/L氢氧化钠溶液10 mL,摇匀,加入转子,连接电位滴定装置,用0.02 mol/L的硫酸滴定,以指示电极的电位变化(△E)为纵坐标,以滴定液体积变化(△V)为横坐标,绘制滴定曲线,以滴定曲线的陡然上升或下降部分的拐点为滴定终点,记录消耗硫酸的体积。依据消耗硫酸的体积折算成消耗氢氧化钠的体积,最终得到提取液中有机酸所消耗氢氧化钠滴定液(0.1 mol/L)体积。每1 mL氢氧化钠滴定液(0.1 mol/L)相当于6.404 mg的枸橼酸(C6H8O7),以枸橼酸(C6H8O7)计计算提取液中总有机酸含量。
2.3 水提取液中总多糖含量测定方法
2.3.1 葡萄糖对照品溶液制备 精密称取无水葡萄糖对照品12.9 mg,置10 mL量瓶中,加蒸馏水适量使溶解并稀释至刻度,摇匀,作为葡萄糖对照品储备溶液(浓度为1.29 mg/mL)。精密吸取对照品储备溶液5 mL,置50 mL量瓶中,加蒸馏水稀释至刻度,摇匀,作为葡萄糖对照品溶液(浓度为0.129 mg/mL)。
2.3.2 苯酚试液配制 取苯酚100 g,加铝片0.1 g,碳酸氢钠0.5 g,蒸馏,收集182 ℃时的馏分10 g,加蒸馏水200 mL使溶解,置棕色瓶中,即得。
2.3.3 最大吸收波长测定 精密吸取葡萄糖对照品溶液(0.129 mg/mL)适量,置具塞试管中,加蒸馏水稀释至2.0 mL,加入苯酚试液1.0 mL,摇匀,迅速滴加浓硫酸5.0 mL,摇匀后放置10 min,置40 ℃水浴中保温15 min,取出,迅速冷却至室温。另取蒸馏水2.0 mL,同法制得空白对照液,于紫外可见分光光度计400~600 nm范围内扫描,其吸收见图3,从吸收图可以看出,葡萄糖经苯酚-硫酸显色后,在490 nm有特征吸收峰。
2.3.4 标准曲线制备 精密吸取葡萄糖对照品溶液(0.129 mg/mL)0.2、0.4、0.6、0.8、1.0 mL(葡萄糖对照品含量分别为0.0258、0.0516、0.0774、0.1032和0.1290 mg),分别加蒸馏水稀释至2.0 mL,按“2.3.3”项下实验,采用分光光度法于490 nm波长处测定吸收度,葡萄糖对照品含量和吸收度对应关系见表3。

图3 葡萄糖对照品紫外扫描谱图
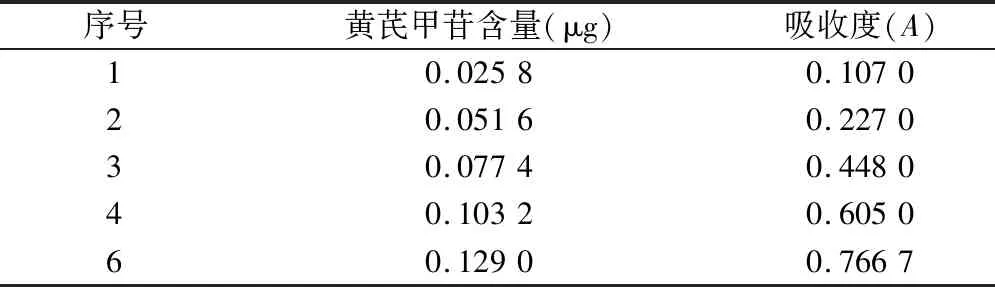
表3 葡萄糖对照品含量与吸收度对应关系
以葡萄糖含量为纵坐标,吸收度为横坐标,用最小二乘法绘制标准曲线,标准曲线方程为:Y=0.151 1X+0.012 3,R2=0.997。线性范围为:0.025 8~0.129 0 mg。
2.3.5 水提取液中多糖含量测定供试品溶液制备 根据相关资料报道,大分子多糖对人体的作用不明显,而小分子多糖具有较强的生理活性,因此在提取液多糖含量测定中,弃去30%乙醇醇沉的大分子多糖类化合物及部分杂质,而对药理作用更明确的小分子多糖进行含量测定。精密吸取正交实验各提取液2 mL,置10 mL试管中,加60%乙醇2 mL,在涡旋混合器上混合60 s,3 000 r/min离心5 min,取出,仔细倾取上清液,沉淀中再加30%乙醇2 mL,同上操作,反复2次,合并上清液,置于10 mL量瓶中,30%乙醇稀释至刻度,摇匀,精密吸取2 mL至10 mL试管中,加无水乙醇5 mL,在涡旋混合器上混合60 s,3 000 r/min离心分离5 min,取出,倾弃上清液,沉淀再加无水乙醇5 mL,同上操作,反复2次,沉淀用水溶解定量转移至5 mL量瓶中,蒸馏水稀释至刻度,摇匀,即得。
2.3.6 供试品最大吸收波长测定 精密吸取水提取液总多糖供试品溶液0.2 mL,置10 mL具塞试管中,按“4.3.3”项下实验。Agilent8453 紫外分光光度计于400~600 nm范围内扫描,其吸收见图4。从图4可以看出,供试品溶液吸收图谱与对照品吸收图谱基本一致。
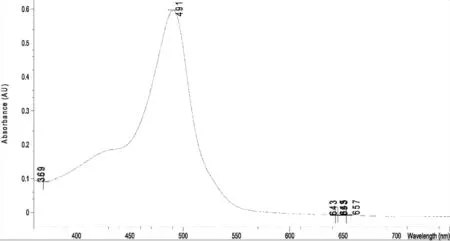
图4 供试液(总多糖)紫外扫描图谱
2.3.7 稳定性考察 精密吸取预实验水提取液2 mL,按“2.3.5”项下操作,制备供试品溶液,再按“2.3.3”项下实验,每隔1 h测定吸收度,测定6次。结果见表4。

表4 稳定性试验结果
从上述稳定性试验结果可以看出,供试液经苯酚-硫酸显色后,供试品溶液在0~2 h内稳定,因此,供试液显色后要在2 h内测定完成。
2.3.8 重复性试验 精密吸取预实验水提取液2 mL,平行6份,分别按“2.3.5”项下操作,制备供试品溶液,再按“2.3.3”项下实验,测定490 nm波长处吸收度,代入曲线方程,计算提取液中总多糖含量。结果见表5。
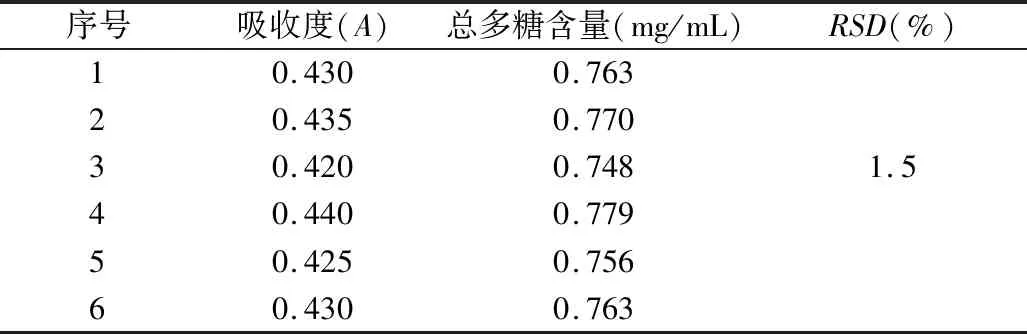
表5 多糖含量测定重现性试验结果
从上述多糖含量测定重现性试验结果可以看出,本含量测定方法重现性良好。
2.4 总皂苷含量测定方法研究
2.4.1 人参皂苷Rg1对照品溶液制备 精密称取105 ℃干燥至恒重的人参皂苷Rg1对照品11.6 mg,置5 mL容量瓶中,加甲醇适量使溶解并稀释至刻度,摇匀,即得(浓度为2.32 mg/mL)。
2.4.2 1%香草醛-高氯酸溶液制备 称取香草醛1.0 g,加高氯酸100 mL使溶解,置棕色瓶中保存。
2.4.3 人参皂苷Rg1对照品显色溶液最大吸收波长测定 精密吸取人参皂苷Rg1对照品溶液(2.32 mg/mL)50 μL,置具塞试管中,于水浴上挥干甲醇,加入1%香草醛-高氯酸溶液0.5 mL,摇匀,60 ℃水浴保温15 min,取出,流水冷却5 min,加入5.0 mL冰醋酸,涡旋混匀。另取甲醇0.5 mL,同法制得空白对照液,紫外可见分光光度计于400~800 nm范围内扫描,其吸收图见图5,从紫外吸收图可以看出,人参皂苷Rg1经香草醛-高氯酸显色后,在544 nm有特征吸收峰。
2.4.4 人参皂苷Rg1对照品标准曲线制备 精密吸取人参皂苷Rg1对照品溶液(2.32 mg/mL)20、30、40、50、70和90 μL,按“4.4.3”项下实验,采用分光光度法于544 nm波长处测定吸收度,吸收度和人参皂苷Rg1对照品含量对应关系见表6。

图5 人参皂苷Rg1对照品紫外扫描图谱
以吸收度为横坐标,人参皂苷Rg1含量为纵坐标,用最小二乘法绘制标准曲线,曲线方程为:Y=0.237X+0.000 2,R2=0.996,线性范围:0.046 4~0.208 8 μg。
2.4.5 提取液中总皂苷含量测定供试品溶液的制备 按水提取液黄芪甲苷供试品溶液制备方法制备总皂苷供试品溶液。
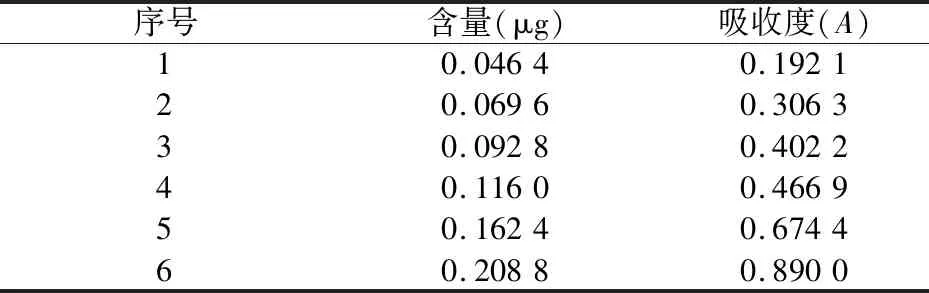
表6 人参皂苷Rg1对照品含量与吸收度对应关系
2.4.6 供试品显色溶液最大吸收波长测定 精密吸取供试品溶液0.2 mL,置10 mL具塞试管中,按“2.4.3”项下实验。紫外分光光度计于400~800 nm扫描,其吸收图谱见图6,从图6可以看出,供试品溶液吸收图谱与对照品吸收图谱最大吸收波长基本一致。

图6 供试液(总皂苷)紫外扫描图谱
2.4.7 供试品显色溶液稳定性考察 精密吸取预实验提取液,按“2.4.5”项下制得供试品溶液,再按“2.4.3”项下实验,每隔1 h测定吸收度,测定6次。结果见表7。

表7 总皂苷供试品溶液显色稳定性试验结果
从上述稳定性试验结果可以看出,供试液经香草醛-高氯酸显色后,样品溶液在0~2 h内稳定,因此,供试液显色后要在2 h内测定完成。
2.4.8 总皂苷含量测定方法重现性实验 精密吸取预实验提取液25 mL,共计6份,分别按“2.4.5”项下平行实验,制备出供试品溶液,再按“2.4.3”项下实验,测定544 nm波长处吸收度。试验结果见表8。
从上述总皂苷含量测定试验结果可以看出,本含量测定方法重现性良好。
2.5 提取工艺优选
2.5.1 正交试验因素水平确定 依据水提取常规,选择对提取效果有影响的加水量、提取时间、药材浸泡时间、提取次数为考察因素,每个因素选择3个常规水平,按L9(3)4正交表进行试验设计。正交试验因素水平见表9。
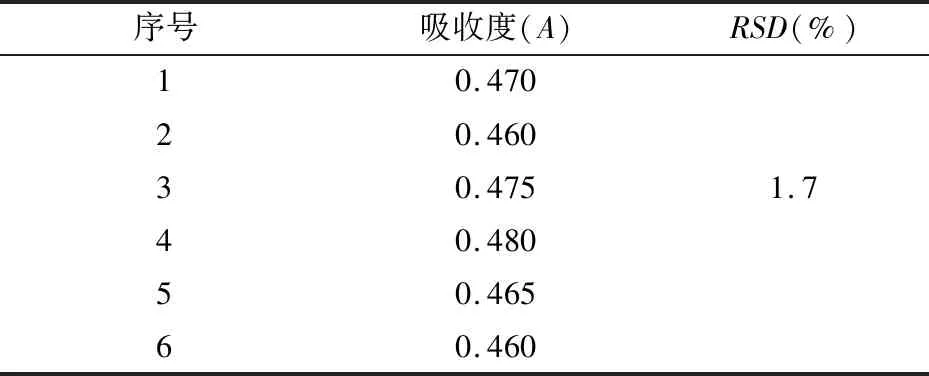
表8 重现性试验结果

表9 水提取正交实验因素水平
2.5.2 正交试验方法及实验安排表 按处方配比,分别取黄芪、麦冬、红参、茯苓、甘草、法半夏、乌梅等13味药材,共计18份,分别混匀,每份总生药量560 g。按表L9(3)4正交试验安排表,进行水提取正交试验,提取液滤过,合并滤液,分别测定各次试验水提取液中黄芪甲苷、总多糖、总有机酸、总皂苷含量。含量测定结果见表10。

表10 各成分含量测定结果
2.6 试验结果分析
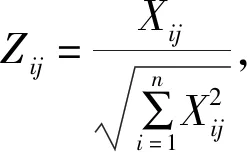
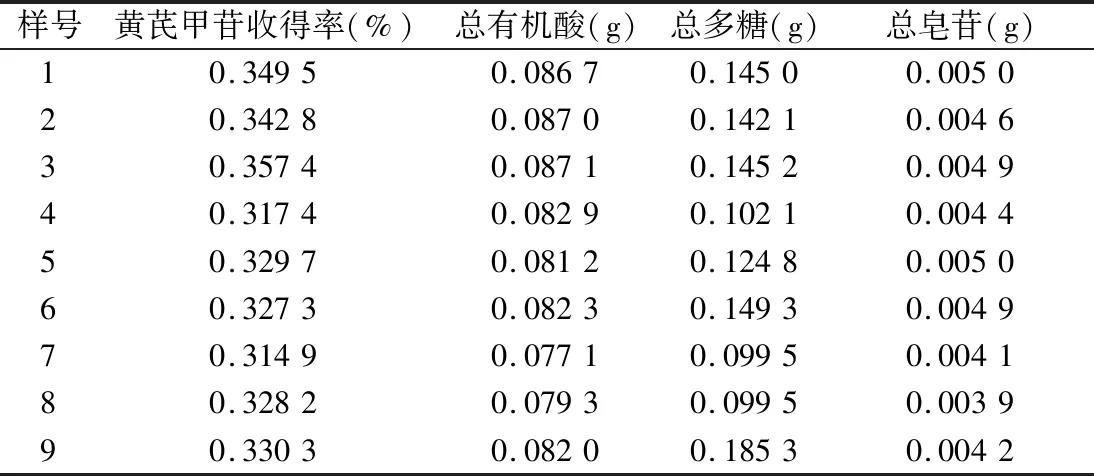
表11 归一化处理矩阵
(2)最优方案与最劣方案。根据归一化理后的矩阵得到最优值和最劣值向量,最优方案:Z+=(0.357 4,0.087 1,0.185 3,0.005 0);最劣方案:Z-=(0.314 9,0.077 1,0.099 5,0.003 9)。
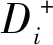
第i个评价对象与最优方案的贴近度Ci(值越大综合效益越好)为:
2.6.2 不同工艺贴近度分析 对化胃舒水提取黄芪甲苷收率、总有机酸含量、总多糖含量及总皂苷含量多工艺目标综合分析,分析结果见表13。

表12 试验方案的欧氏距离和贴近度
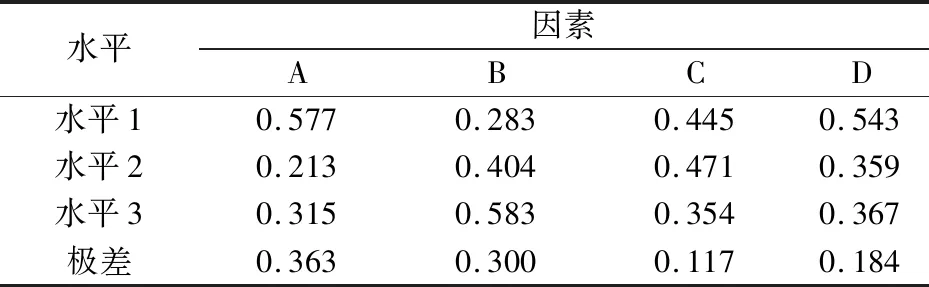
表13 工艺参数各水平综合平均贴近度
结果表明,对水提取工艺影响因素数排序为A>B>D>C,即加水倍数和提取时间的影响较大,浸泡时间和提取次数影响较小,最佳提取参数为A1B3C2D1。
2.7 工艺验证
参为验证工艺的稳定性,用最佳提取工艺进行3批验证试验,结果见表14。结果显示3批样品的黄芪甲苷收率、总有机酸、总多糖、总皂苷的平均值分别为91.27%、23.48 g、40.69 g、1.35 g,其RSD分别为1.2%、1.2%、1.8%、2.7%,表明工艺稳定可行。

表14 验证试验结果
3 讨论
目前,测定植物中多糖含量主要是利用多糖在浓硫酸作用下迅速脱水成糠醛或羟甲基糠醛,再由酚类、芳胺类等检测剂与之反应,生成具有特定颜色的有机化合物,在一定范围内,溶液颜色的深浅与糖的含量成正比[13]。根据显色剂的不同,已知显色方法有苯酚-硫酸法[14-15]、蒽酮-硫酸法[16-17]、咔唑-硫酸法[18]等。苯酚-硫酸比色法主要用于甲基化的糖、戊糖、寡糖类以及多聚糖的测定,甚至可以用于糖肽和糖蛋白的测定。此法优势在于可以进行大量样品的测定,实验时基本不受蛋白质存在的影响,且产生的有色化合物在120 min内颜色稳定,因此选择苯酚-硫酸法用于总多糖含量测定。
乌梅及处方中其他药材所含有机酸具有调理胃肠蠕动、促进溃疡性结肠炎组织细胞再生、修复胃黏膜损害、促进胃酸分泌等作用,因此在制备工艺研究中拟对水提取液中总有机酸含量进行测定。现阶段中药提取液中总有机酸含量测定方法主要有化学滴定法及电位滴定法。由于中药提取液颜色深,严重干扰化学滴定法滴定终点判断,因此选择电位滴定法测定水提取液中总有机酸含量。
目前中药提取液中总皂苷含量测定方法主要有重量法及紫外-可见分光光度法,由于复方中含皂甙类化合物药材多,重量法测定实验误差较大,因此本制剂工艺研究水提取液中总皂苷含量测定方法拟首选紫外-可见分光光度法。
TOPSIS通过将多指标计算为一个综合指标,克服人为确定权重系数的片面性及主观性,提高分析方法的可靠性,该方法已应用到多种中药质量评价。如姚入宇等[19]、陈家仪等[20]、李运等[21]、罗益远等[22],分别采用Topsis法对北柴胡种子、猴耳环、三七、桔梗药材的质量进行了评价;刘书斌等[23]基于AHP法优化的熵权TOPSIS模型对不同产地黄花菜药材质量的进行了综合评价;马天翔等[24]基于OPLS结合熵权TOPSIS法对不同产地锁阳进行了鉴别与综合质量评价;严倩茹等[25]、张娜等[26]将TOPSIS与灰色关联分析相结合分别对维C银翘片及不同产地广西郁金的质量进行了评价。本研究通过测定化胃舒水提取液中黄芪甲苷收率、总有机酸含量、总多糖含量、总皂苷含量,与 TOPSIS综合评价法相结合,计算不同提取条件的化胃舒水提取液各指标成分的综合评价数值,其结果更具有客观性和科学性,最终优选出化胃舒颗粒水提取的最佳工艺条件,即浸泡2 h,加水提取2次,每次加水10倍量,提取2 h。
猜你喜欢
浙江中西医结合杂志(2022年10期)2022-10-25
中草药(2022年19期)2022-10-14
广州中医药大学学报(2022年4期)2022-03-29
农产品加工(2022年4期)2022-03-11
毛纺科技(2021年8期)2021-10-14
昆明医科大学学报(2021年6期)2021-07-31
中华胰腺病杂志(2021年2期)2021-04-26
中国土壤与肥料(2020年1期)2020-12-22
安徽中医药大学学报(2020年2期)2020-04-21
医学新知(2019年4期)2020-01-02
