
氮磷比对脂多糖胺/质粒复合物性能的影响*
2022-11-24 09:47关云高彦晨张兴滕伟
广东医学 2022年10期
关云, 高彦晨, 张兴, 滕伟
1广州中医药大学第二附属医院、广东省中医院口腔科(广东广州 510120);2中山大学光华口腔医学院、中山大学附属口腔医院修复科、中山大学口腔医学研究所(广东广州 510055)
细胞表面的糖蛋白和糖脂存在含负电的唾液酸残基,裸基因表面呈多阴离子电性,基因自发进入体细胞是一个低效的过程[1-2],因此,阳离子脂质介导的基因转移已成为研究最广泛和最常用的非病毒基因传递方法[3]。以聚乙烯亚胺(polyethylenimine,PEI)为代表的阳离子聚合物基因载体转染效率与N/P(PEI中的氨基与DNA磷磷酸基团的摩尔比值)有关,N/P越高,载体结合、压缩DNA的能力越强,形成的复合物的粒径越小,利于细胞胞吞,进而有利于提高转染效率[4]。然而N/P的增加也会带来更强的正电性,导致细胞毒性增加,且不利于入胞后DNA的释放,进而影响转染效率[5]。我们采用了一种新型的非病毒载体,由低分子量1.8 kD的PEI、疏水性的胆固醇(cholesterol, Cho)和亲水性良好的阴离子氧化海藻酸钠(anionic oxidized sodium alginate, OA)合成,新合成的阳离子聚合物叫做海藻酸钠-聚乙烯亚胺-胆固醇嵌段共聚物脂多糖胺(lipopolysaccharide-amine, LPSA)。合成的LPSA可溶于水,并能在水中通过静电作用形成可溶于水的纳米囊泡载体。在口腔临床工作中,由于遗传因素、肿瘤、外伤等引起的,先天性或获得性的颌骨缺损十分常见。2019年2—10月课题组应用新型的LPSA阳离子脂质纳米颗粒,与骨形态生成蛋白2(bone morphogenetic protein 2, BMP2)质粒(后文简称为pDNA)结合,以期构成LPSA/pDNA复合物,为后续成骨诱导的相关研究奠定基础。本研究探讨不同N/P时,新型非病毒载体LPSA与pDNA构建的复合物理化性能的变化。
1 材料与方法
1.1 主要试剂及仪器 非融合表达的人源性真核表达质粒(产品编号:EX-A0241-M61;Gene-Copoeia公司,美国);快速滤过法无内毒素质粒大提试剂盒(OMEGA公司,美国);紫外分光光度计(Thermo公司,美国),支链型聚乙烯亚胺(PEI;相对分子质量1.8×103Da)、PEI 25 kD(相对分子质量25×103Da)(Sigma公司,美国);LPSA(由中山大学心血管循环辅助实验室合成并惠赠);Lipofectamine 2000(Sigma公司,美国);原子力显微镜(AFM, Shimadzu SPM-9500J3);纳米粒度及电位分析仪Zeta sizer Nano-ZS90 Nano series(Malvern Instruments公司,英国);氨苄青霉素钠(MDBio Inc,中国);透射电镜(JEOL公司,日本);动态光散射(dynamiclight scattering, DLS)仪(Malvern公司,英国)。
1.2 质粒扩增与检测 质粒扩增:配置溶菌肉汤(lysogeny broth, LB)培养基,用平板划线法,挑取pDNA大肠杆菌(GeneCopoeia公司,美国)单克隆菌落,接种于氨苄青霉素钠浓度为30 μg/mL的溶菌肉汤培养液中,37℃摇床培养16 h,用Fastfilter Endo-Free Plasmid Maxi Kit(OMEGA公司,美国)提取、纯化质粒。DNA浓度采用紫外分光光度计(Thermo公司,美国)在260 nm紫外吸收法测定。用PBS(pH 7.4)分别将质粒DNA稀释至40 mg/mL,保存于-80℃备用。
质粒检测:配制0.9%的琼脂糖凝胶,置于TBE缓冲液中,将pDNA稀释至40 ng/μL,37℃分别进行Xho Ⅰ单酶切及Xho Ⅰ+Hind Ⅲ双酶切2 h,与上样缓冲液混合均匀后,110 V电泳0.5 h,紫外线照射下,拍摄图片。
1.3 LPSA/pDNA复合物构建 用无菌超纯水将LPSA(1.85 mg/mL)稀释成所需浓度,与pDNA(40 mg/mL)溶液等体积混合,轻轻吹打并涡旋混合3~5 s,25℃静置30 min后立即使用。
1.4 LPSA/pDNA阻滞能力与N/P的关系 分别配制N/P(LPSA中的氨基与DNA磷酸基团的摩尔比)为0(裸DNA)、2、4、8、10、60的LPSA/pDNA溶液;设置阴性对照PEI 25k/pDNA复合物(N/P为2)。制备0.9%的琼脂糖凝胶(含0.1 μg/mL溴化乙锭)取10 μL样本与2 μL上样缓冲液混合均匀后,置于Tris-boric acid-EDTA(TBE)缓冲液中,上样,110 V电泳0.5 h,紫外线显影。
1.5 LPSA/pDNA粒径及zeta电位与N/P变化关系 25℃,按前述步骤分别配制N/P为0、1.5、3、6、12、25、60的LPSA/pDNA溶液1.5~2 mL,置于Nano-ZS90型粒径分析仪测定复合物的粒径和ζ-电位。
1.6 LPSA/pDNA形貌观察 形貌观察:配制0.5 mg/mL的LPSA和LPSA/pDNA(N/P为60)各50 μL,25℃磷钨酸负染30 min后,透射电镜下观察LPSA和LPSA/pDNA形态并测量直径。
1.7 LPSA/pDNA体外耐酶降解实验 配制N/P为60的LPSA/pDNA溶液10 μL,与等体积1 U的DNase Ⅰ混合均匀,37℃分别放置1、2、4、6 h后,加入5 μL EDTA(100 mmol/mL)终止酶解反应,37℃水浴10 min后,加入10 μL肝素(12 mg/mL),25℃放置2 h置换DNA,取样行凝胶电泳分析。阳性对照组为LPSA/pDNA未加DNase Ⅰ组;阴性对照组为pDNA加DNase Ⅰ孵育15 min。制备0.9%的琼脂糖凝胶(含0.1 μg/mL溴化乙锭)取10 μL样本与2 μL上样缓冲液混合均匀后,置于Tris-boric acid-EDTA(TBE)缓冲液中,上样,110 V电泳0.5 h,紫外线显影[6]。
2 结果
2.1 质粒制备 pDNA提纯浓度为805.37 ng/μL。OD260/280为1.86,介于1.8~1.9之间,表明pDNA所含蛋白质及RNA成分较少;OD260/230为2.14,接近正常范围(2.0~2.5)下限,表明pDNA中小分子及盐类物质含量较少,证实所获pDNA纯度较高。
pDNA酶切实验结果示:Xho Ⅰ单酶切后,位于DNA marker约6 500+ bp处可见荧光条带,Xho Ⅰ+Hind Ⅲ双酶切后,1 000 bp marker条带附近可见插入为人源性BMP2基因片段,大小为1 191 bp(图1)。pDNA质粒大小为7 748 bp(图1)。
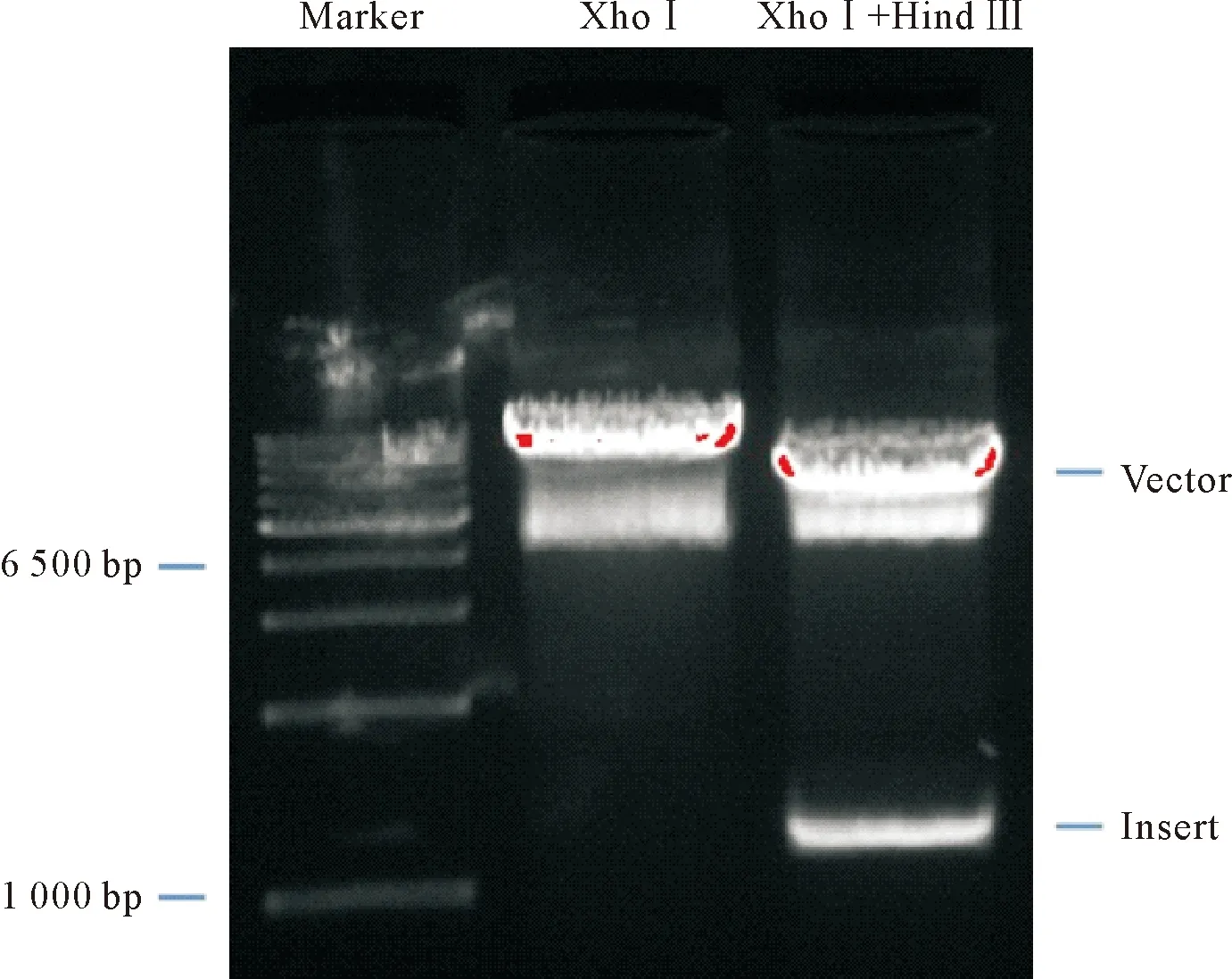
图1 限制性内切酶验证插入BMP2基因片段大小
2.2 LPSA/pDNA阻滞能力与N/P的关系 随着N/P增加,当N/P>4时,琼脂糖凝胶电泳条带荧光显色逐渐变弱,阻滞在上样孔中的DNA亮度增加,最终与阴性对照PEI 25k/pDNA一致;阴性对照组PEI 25k/pDNA只在上样孔中可见DNA条带荧光。表明,随着N/P增加,LPSA/pDNA阻滞质粒DNA能力增加;当N/P>10时,LPSA可基本阻滞pDNA(图2)。

注:A~G, LPSA/pDNA的N/P依次为:0,1,2,4,8,10,60;H, PEI 25k/pDNA
2.3 LPSA/pDNA粒径及zeta电位与N/P变化关系 LPSA/pDNA复合物的粒径随着N/P比的增加先减小后增大,当N/P>3时,LPSA/pDNA粒径<150 nm;当N/P为12~25时,LPSA/pDNA粒径稳定于65 nm左右,LPSA/pDNA表面zeta电位呈正电性,电荷值为9~35 mV。见表1。

表1 LPSA/pDNA粒径及zeta电位随N/P增加的变化
2.4 LPSA/pDNA的形貌观察 透射电镜下可见LPSA呈类球形、椭球形,直径约10~25 nm(图3-A);LPSA/pDNA呈球形或椭球形空心囊泡状,直径约40~120 nm(图3-B、C)。

注:A: LPSA; B、C: LPSA/pDNA
2.5 LPSA/pDNA抗酶解能力实验 当N/P为60时,随DNaseⅠ处理时间增加,4 h内琼脂糖凝胶电泳条带亮度无明显变化(图4-C~E),与阳性对照组(图4-A)LPSA/pDNA条带一致;6 h时琼脂糖凝胶电泳条带亮度稍微减弱(图4-F);阴性对照组pDNA与DNaseⅠ酶混合孵育6 h,可见B泳道模糊小分子DNA条带泳出(图4-B);阳性对照组LPSA/pDNA未加DNaseⅠ酶孵育6 h仍可见清晰条带。因此,在4 h内LPSA/pDNA抗DNaseⅠ酶解,6 h 时LPSA对pDNA保护能力稍有下降(图4),见图5。

注:A:LPSA/pDNA无DNaseⅠ在25℃孵育6 h;B:pDNA与DNaseⅠ混合6 h;C~F:LPSA/pDNA与DNaseⅠ混合时间依次为:1 h、2 h、4 h、6 h

图5 LPSA/pDNA复合物粒径及Zeta电位随N/P比变化关系
3 讨论
PEI是研究最深入的非病毒基因载体之一[7]。大量研究表明,分子量影响PEI的转染效率和细胞毒性,这与PEI带正电荷的胺基数量有关,因此,随着分子量的增加PEI毒性也随之增加。高分子量PEI(如PEI 25 kD)具有较高的转染效率和转染效率高细胞毒性,低分子量PEI(如PEI 2 000 Da)具有低功率细胞毒性和较低的转染效率[8],其DNA浓缩能力也相应减弱[9]。各国学者通过在PEI上接枝生物可降解的链段,如:氧化海藻酸盐、聚乙二醇及其衍生物、氨基酸片段等[10],希望达到提高转染效率并降低毒性的目的。曾有学者[8]将胆固醇接枝到PEI(1.8 kD)上,通过胆固醇受体通路介导的胞吞作用,提高复合物入胞能力,进而提高转染效率。经胆固醇修饰后的载体既具有亲水基团有具有疏水基团,在提高基因载体的血清稳定性的同时,可使载体/pDNA复合物免受血清中脂肪酶、核酸酶和高密度脂蛋白的降解[9]。
LPSA通过在PEI(1.8 kD)上接枝Cho,形成疏水侧链;带负电荷的OA作为主链,与PEI接枝形成亲水基团,形成三嵌段两亲两性脂质共聚物LPSA(OA-PEI-Cho)。选择低分子量PEI是在最大程度降低共聚物毒性,保留其正电性及PEI固有的“质子海绵效应”[7, 10]。Cho作为侧链的阳离子基因载体可有效提高了载体的膜流动性、稳定性;有效促进基因药物的释放并降低毒性[9];具有较强的DNA、siRNA结合能力[8];疏水基团的引入能帮助LPSA在水溶液或体内环境形成纳米粒子结构,并介导LPSA通过胆固醇受体模式进行胞吞,提高入胞效率,减少与细胞膜的接触时间,进而达到降低毒性并提高转染效率的目标。OA由带阴离子醛糖环组成,用以屏蔽PEI的部分正电荷,降低载体毒性;OA醛糖环之间链接的糖苷键在体内环境下可断裂进而降解,使其具有良好的生物相容性和可降解性。我们期望OA的引入,能克服阳离子脂质体与表面电荷有关的缺点,如毒性和非特异性电荷干扰等[11];能有效降低LPSA的表面正电荷,增加复合物稳定性,具备阴离子特性,良好的生物相容性和体内可降解性,最终能达到降低 LPSA 的细胞毒性的目的。
LPSA与DNA的结合能力,是评价其作为基因载体介导基因传递的先决条件,LPSA在合成过程中引入的Cho和OA降低了伯胺的密度,在一定程度上降低了其DNA结合能力,但是因为LPSA作为阳离子聚合物,具有亲水核和被亲水电晕包围的封闭疏水膜,这一特有的纳米囊泡结构与其他具有固体核的纳米载体不同,增加了载体表面可用的伯胺数量,能补偿引入Cho和OA对LPSA的 DNA结合能力的影响。通过DNA凝胶电泳阻滞实验不难看出,在N/P为4~60时,上样孔中的荧光条带亮度增强,可自由泳出的DNA条带亮度减弱(图1)。说明,随着N/P增加,底物中LPSA所占比例不断增加,形成的LPSA/pDNA复合物随之增加,而复合物在凝胶中的泳动速率明显低于DNA分子,从而,阻滞在上样孔中的DNA量也随之增多。当N/P为10~60时,基本仅在上样孔中看到荧光条带,泳道中DNA条带荧光基本消失,上样孔条带亮度与对照组PEI 25k/pDNA电泳图像一致(图1)。意味着,当N/P>10时,底物中的LPSA已基本将DNA结合并阻滞在上样孔中,形成LPSA/pDNA复合物能明显阻滞 DNA移动。由此可知,当N/P>10时,LPSA可基本阻滞pDNA,有良好的DNA结合能力。
复合物粒径大小与细胞内吞过程有关,粒径过大不利于细胞对复合物分子进行胞吞,从而影响转染效率[12]。基因转染的过程中,通常要求复合物粒径<150 nm[13-15]。LPSA/pDNA复合物的粒径随着N/P比的增加先减小后增大,当N/P>3时,LPSA/pDNA复合物的粒径约为均<150 nm,最小粒径接近65 nm,满足基因转染载体的粒径要求[6, 16]。图5可看出,随着N/P的不断增加,LPSA/pDNA复合物粒径先减小后缓慢增大,在N/P为12~25时,复合物粒径趋于稳定,约65 nm。随着N/P的增加,LPSA作为阳离子基因载体对DNA的缩聚作用不断增大,LPSA/pDNA复合物粒径呈不断减小的趋势,此时复合物内的缩聚作用占主导;随着N/P进一步增大,阳离子聚合物本身的正电性不断增大,随着电荷密度增大,LPSA/pDNA复合物粒径呈缓慢增大的趋势。
许多阳离子基因载体,在体外表现出很高的效率,但其表面通常有较高的电荷密度,因此在具有高转染力的同时具有较强的细胞毒性。从文献可知,阳离子聚合物基因载体的Zeta电位会对其基因负载能力和转染效率产生很大影响[16-18]。复合物表面电荷会影响复合物被细胞胞吞的过程,从而进一步影响载体的毒性及转染效率。不同N/P对LPSA/pDNA复合物Zeta电位的影响列于表1,复合物表面始终呈正电性,间接证明了LPSA/pDNA表面是以带正电性的基团为外冠的,对细胞表面吸附及胞吞具有积极的促进作用。当N/P为1.5~25时,Zeta电位在一个较小的范围(9.95~17.00 mV)波动;随着N/P进一步增加,Zeta电位有进一步增大的趋势。由此可推测,pDNA表面所带负电荷先被LPSA中和,电荷排斥作用减弱,并达到复合物缩聚的效果,随着LPSA的增加,复合物表面的正电性增强。
阳离子聚合物/DNA复合物的表面电荷对其电稳定性和对负电荷细胞膜的亲和力非常重要。由实验结果可知,在N/P>10时,LPSA与质粒可完全复合,当N/P<25时,Zeta电位稳定在20 mV以内,呈弱正电性,可极大降低其细胞毒性[16, 18-19]。以PEI为代表的阳离子聚合物/基因复合物随着 N/P升高,载体结合、压缩 DNA的能力越强,转染效率转染效率越高[7];而复合物的正电性亦会越强,导致细胞毒性增加,且不利于入胞后 DNA的释放,进而影响转染效率。在尽可能兼顾弱正电性及低细胞毒性的基础上,我们选择N/P为60的LPSA/pDNA作为进一步观察对象,此时,其粒径大小符合基因转染的需求,约为112 nm,LPSA 能够较好地包裹 DNA 并形成粒径较小的纳米粒子。
在水溶液中,LPSA形成囊泡依赖于两个主动驱动力,一是疏水基团Cho的相互作用,另一则是PEI与OA的静电相互作用。结果(图3)显示:LPSA 与LPSA/pDNA复合物均以类球形或坍塌的足球形状存在,而不是一个充盈的球体。这与透射电镜制样、观察过程有关。因为透射电镜样品需要在真空无水条件下进行观察,观察过程中水分的蒸发使得纳米球内部中空,纳米球囊塌陷,形成了内陷囊泡状的形貌。然而动态光散射粒径仪是在水溶液中进行,并且DLS测定的结果是对溶液内所有纳米粒径进行测量,取正态分布平均值,可对LPSA与LPSA/pDNA粒径进行更准确的评估。
纳米阳离子聚合物载体在实现基因传递的过程中,克服了与细胞表面的结合及内吞作用等重重困难之后,则需要逃避溶酶体的消化破坏[6, 19]。溶酶体逃逸[20]后,所载目的基因或药物才有机会释放到胞质中发挥其功能。耐酶解试验中,证实4h内,LPSA对pDNA分子具有耐酶解保护力,为躲避细胞内的酶解反应,实现基因传递入核奠定了基础[6]。
大量阳离子基因载体转染试验表明:在阳离子/DNA复合物中N/P比越大,基因转染效率越高,但细胞毒性也越强[21]。细胞毒性的大小与复合物入胞时的理化状态密不可分,本研究探究了LPSA与pDNA在何种N/P条件下,形成的LPSA/pDNA复合物的理化性能可达到最佳平衡点(最佳N/P比)[5, 22],为进一步探讨其作为高效基因载体的可行性奠定基础。本研究以PEI 25 kD作为阳性对照,将携带BMP2基因的质粒和LPSA缩合成不同N/P比的复合物,探究N/P对LPSA/pDNA复合物理化性能的影响,就LPSA加载DNA的最佳N/P比进行初步探讨。证实:N/P为12~25时,LPSA与pDNA形成直径约65 nm的阳离子复合物纳米囊泡。N/P为60时,LPSA可有效结合pDNA,LPSA/pDNA在4 h内,耐DNaseⅠ酶解。LPSA在基因递送治疗方面有广阔的应用前景。
利益相关声明:所有作者共同认可论文无利益冲突。
作者贡献说明:关云直接参与实施研究,采集数据,分析/解释数据,起草论文,统计分析,支持性贡献。高彦晨采集数据,起草论文,统计分析,行政、技术或材料支持,支持性贡献。张兴对论文的知识性内容作批评性审阅,行政、技术或材料支持,支持性贡献。滕伟酝酿和设计实验,分析/解释数据,对文章的知识性内容作批评性审阅,获取研究经费,行政、技术或材料支持,指导,支持性贡献。
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02
中国化妆品(2022年2期)2022-03-04
昆明医科大学学报(2021年3期)2021-07-22
西华大学学报(自然科学版)(2020年6期)2020-10-15
有色金属材料与工程(2018年1期)2018-11-25
安徽化工(2018年4期)2018-09-03
西部论丛(2017年5期)2017-10-25
商情(2017年30期)2017-09-18
江苏农业科学(2016年1期)2017-05-17
药学研究(2015年11期)2015-12-19
