
先天性虹膜缺损合并先天性白内障一家系的PAX6基因新突变
2022-11-18 09:49:48顾静易浩安查旭孔艳波江伟阳杨芳李凡何永蜀
中华实验眼科杂志 2022年10期
顾静 易浩安 查旭 孔艳波 江伟阳 杨芳 李凡 何永蜀
1云南省残疾人康复中心眼科,昆明 650032;2昆明医科大学细胞生物学与医学遗传学系,昆明 650500;3昆明医科大学第二附属医院眼科,昆明 650101;4昆明医科大学病理学与病理生理学系,昆明 650500
先天性虹膜缺损或无虹膜是一种罕见的常染色体显性遗传性眼组织畸形,其在中国的发病率约为1∶ 100 000[1],以部分或全部虹膜缺损为主要临床表现,同时可伴有其他眼部结构异常,包括角膜混浊、青光眼、白内障、晶状体异位、斜视和眼球震颤等[2]。约2/3的虹膜缺损患者是由于家族遗传引起,剩余的散发病例由新发生的基因突变引起[3]。先天性虹膜缺损发病无明显的种族区别。人配对盒基因6(paired box gene,PAX6)是脊椎动物PAX基因家族成员之一,在神经组织,尤其在眼的发育中起着重要作用[4],至今人类基因组突变数据库已收集592种PAX6基因突变(http://www.hgmd.cf.ac.uk/ac/gene.php?gene=PAX6),其中92%的突变与虹膜缺损或无虹膜有关。PAX6蛋白是一个高度保守的转录调节蛋白,该蛋白通过与其配对的DNA结合域识别靶基因,从而调控下游靶基因的转录[5]。PAX6基因突变占先天性虹膜缺损病因的90%以上,其变异方式多样,有基因缺失、无义突变、移码突变、剪切位点突变和错义突变等[6]。除PAX6基因以外,其他与先天性虹膜缺损伴眼部疾病有关的基因包括ITPR1、ELP4、TRIM44、FOXE3、FOXC1和PITX2等[6-8]。本研究采用全外显子测序和生物信息学分析方法对云南省先天性虹膜缺损合并先天性白内障一家系进行致病基因分析,探讨该家系发病的遗传学基础。
1 资料与方法
1.1 一般资料
采用家系调查研究方法,2020年2月于云南省残疾人康复中心和昆明医科大学第二附属医院眼科收集云南汉族先天性虹膜缺损合并先天性白内障一家系。该家系3代共8名成员,包括3例患者和5名表型正常者。本研究遵循《赫尔辛基宣言》,研究方案经昆明医科大学第二附属医院伦理委员会批准(批文号:审-PJ-2020-61),所有参与研究的患者及家属均对本研究目的知情并自愿签署知情同意书。
1.2 方法
1.2.1一般检查 对先证者及其父母、子女和丈夫进行视力、验光、角膜曲率检查(ARK-1自动电脑验光/角膜曲率仪,日本NIDEK公司);采用裂隙灯显微镜(YZ5E,苏州六六视觉科技有限公司)进行眼前节检查;采用眼科A/B型超声诊断仪(ODM_2100S,天津迈达医学科技股份有限公司)测量眼轴长度,并观察玻璃体腔情况;采用眼科超声生物显微镜(MD-300L,天津迈达医学科技股份有限公司)进行房角检查及晶状体厚度测量;采用非接触式眼压计(NT-2000,日本NIDEK公司)测量眼压;采用光相干断层扫描仪(Cirrus HD-OCT MODEL 500,德国Carl Zeiss公司)行视网膜层间结构检查;采用眼底照相机(TRC-NW7SF,日本Topcon公司)观察视网膜情况。对该家系成员进行全身检查,以排查是否伴有其他系统异常。
1.2.2基因检测及数据分析 基因检测和生物信息学分析由昆明医科大学细胞生物学与医学遗传学系完成。采用高通量测序技术对先证者及其丈夫进行全外显子组测序,外显子区域使用Agilent SureSelect Human All Exon V6 kit(Agilent Technologies,Palo Alto,CA,USA)进行捕获。DNA文库采用NovaSeq6000高通量测序系统(Illumina,San Diego,CA,USA)进行双端150 bp测序。原始测序数据使用FASTQC(http://www.bioinformatics.babraham.ac.uk/projects/fastqc)进行质量控制。利用BWA(Burrows-Wheeler Aligner)[9]将reads比对到hg19人类基因组(UCSC human genome hg19 build)。单核苷酸变异和插入/缺失变异由SAMTOOLS[10]识别,并通过ANNOVAR[11]注释,获得变异位点的位置、变异类型、保守性、危害性等信息。并将变异位点比对到dbSNP数据库(https://www.ncbi.nlm.nih.gov/snp)、千人基因组计划数据库(http://www.1000genomes.org)进行筛选。采用homozygositymapper[12]定位常染色体隐性遗传可疑基因座。采用UGENE进行氨基酸保守性分析。采用Mutation Taster[13]预测变异对蛋白翻译的影响。参照美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)遗传变异分类标准与指南对变异位点进行致病性评估。针对筛选出的变异位点经Sanger测序验证,同时在家系成员中验证共分离现象并确定变异位点。
2 结果
2.1 家系患者临床表型

图1 先天性虹膜缺损合并先天性白内障患者家系图 □:正常男性;○:正常女性;■:男性患者;●:女性患者;:先证者Figure 1 Pedigree of the family with congenital iris coloboma and congenital cataract □:normal male;○:normal female;■:male patient;●:female patient;:proband
该家系中第2代和第3代连续出现3例患者,其中男1例,女2例(图1)。先证者Ⅱ-2,女,29岁,双眼虹膜大部分缺损,仅周边部见少量虹膜组织,晶状体皮质及后囊混浊,伴眼球震颤,眼部检查无其他畸形,角膜透明,眼压正常。先证者子女(Ⅲ-1、Ⅲ-2)均表现为先天性虹膜缺损合并先天性白内障症状。先证者之子Ⅲ-1双眼虹膜大部分缺损,仅可见周边部分0.2~1.0 mm虹膜,双眼晶状体皮质混浊累及后极部,并伴眼球震颤,角膜光滑透明,眼部检查无其他畸形,眼压正常。先证者之女Ⅲ-2双眼虹膜大部缺损,仅可见周边部分0.2~1.0 mm虹膜,双眼晶状体后囊轻度混浊,伴眼球震颤,角膜光滑透明,眼压正常。该家系3例患者白内障症状随年龄增长进行性加重,未合并其他并发症,患者临床表现均支持先天性虹膜缺损合并先天性白内障的诊断。先证者丈夫Ⅱ1双眼眼位正,无眼球震颤。双眼角膜、结膜正常,前房中深,巩膜纹理清晰,瞳孔直径2.5 mm,对光反射灵敏,晶状体透明,眼底正常(图2)。该家系遗传方式符合常染色体显性遗传,为先证者新发变异并遗传给下一代。
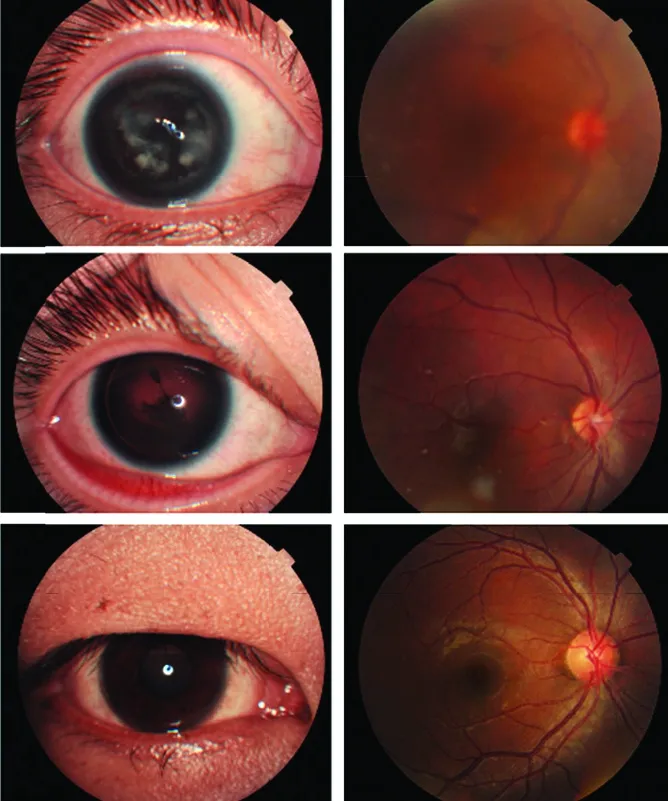
图2 先证者Ⅱ-2及其儿子Ⅲ-1、丈夫Ⅱ-1右眼眼部检查照片 A、C、E:Ⅱ-2、Ⅲ-1、Ⅱ-1眼前节图像 B、D、F:Ⅱ-2、Ⅲ-1、Ⅱ-1眼底图像Figure 2 Right eye images of proband Ⅱ-2,her son Ⅲ-1,and her husband Ⅱ-1 A,C,E:Anterior segment images of Ⅱ-2,Ⅲ-1 and Ⅱ-1 B,D,F:Fundus images of Ⅱ-2,Ⅲ-1 and Ⅱ-1
2.2 家系突变基因检测分析
先证者及其丈夫的外显子区域平均测序深度分别为145.56X和138.28X,平均测序深度>20X的百分比分别为99.0%和98.6%。按常染色体隐性遗传方式分析,根据先证者及其丈夫的变异位点进行homozygositymapper定位分析,一共有32个常染色体隐性遗传可疑基因座被鉴定,将其与先证者的基因组变异信息比较,一共筛选出7个可疑基因,分别为ZNF852、MICA、SLC37A4、VPS11、PTGES3、C14orf180和SPPL2B,但未发现这些基因与眼发育相关的证据和资料,不能支持该病的常染色体隐性遗传方式。按常染色体显性遗传方式进行分析,在先证者PAX6基因第8外显子上发现了1个杂合单碱基插入导致的移码突变c.415dupA(p.R139fs),OMIM数据库、HGMD数据库和ClinVar数据库均未报道该变异。先证者丈夫不存在该变异。MutationTaster预测结果显示,该变异位点位于PAX6蛋白高度保守区,属于移码突变,影响了后续蛋白质的正常翻译。结合本家系特征,认为该家系符合常染色体显性遗传。Sanger测序结果显示,家系中3例患者(先证者及其子女)均携带此移码突变,均为杂合变异,先证者父母及其丈夫不携带此变异,符合家系共分离(图3)。PAX6同源蛋白序列比对结果显示,发生移码突变的位点在各物种间保守(图4)。进一步分析其改变后对氨基酸序列的影响,发现移码突变后仅翻译7个氨基酸就出现了终止密码,此变异对PAX6蛋白一级结构改变巨大(图5)。根据ACMG遗传变异分类标准与指南,移码突变评为PVS1,数据库正常人群对照中未发现该变异评为PM2,符合家系共分离评为PP1,该变异总致病性评分为PVS1+PM2+PP1,为致病性变异。
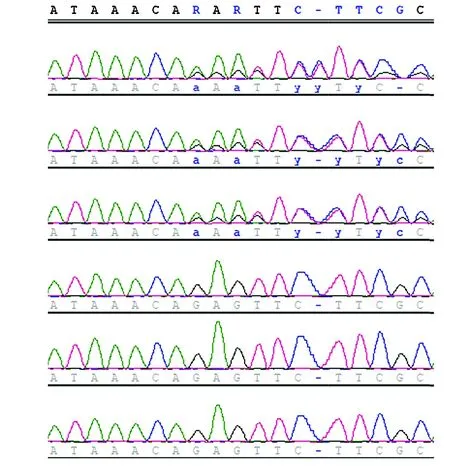
图3 PAX6基因c.415dupA变异Sanger测序图 箭头所示为插入位点Figure 3 Sanger sequencing map of PAX6 c.415dupA variant Arrow showed the insertion site

图4 PAX6同源蛋白序列比对图 PAX6蛋白第139位精氨酸在物种间高度保守Figure 4 Sequence alignment of PAX6 homologous proteins The arginine at position 139 of PAX6 protein was highly conserved among multiple species

图5 c.415dupA(p.R139fs)对PAX6翻译的影响 野生型翻译出完整的PAX6序列,c.415dupA(p.R139fs)在插入位点后翻译7个氨基酸停止翻译Figure 5 Effect of c.415dupA (p.R139fs) on PAX6 translation The wild type normally translated the complete sequence of PAX6,but translation stopped with 7 amino acids translated after the insertion of c.415dupA (p.R139fs)
3 讨论
先天性虹膜缺损是在胚胎发育过程中神经外胚层和中胚层发育障碍导致的眼部结构发育异常[2]。PAX6基因在眼的正常发育中起着极其重要的作用,其位于11号染色体短臂1区3带(11p13),全长22 kb,含14个外显子,第1~3外显子属于非编码区,第4~14外显子编码含436个氨基酸的高度保守的转录因子,通过识别特定的碱基序列与之结合调控下游基因的转录[14-15]。PAX6基因是同源异形基因,其功能特点与基因剂量有关,同源染色体上PAX6等位基因的转录和翻译是眼正常发育的关键;如果其中1个PAX6等位基因由于染色体重排发生缺失或基因突变不能行使其功能,将导致PAX6基因单倍体剂量不足,80%~90%的虹膜缺损或无虹膜与PAX6基因单倍体剂量不足有关[16]。由于PAX6基因的剂量敏感性,单倍体剂量不足将影响人正常视杯视柄的发育,改变虹膜和睫状体组织的生长和分化,出现无虹膜或虹膜缺损症状[17]。
本家系中先证者父母未患病,因此我们首先判断家系遗传方式为常染色体隐性遗传,但进行生物信息学分析后不支持该家系为常染色体隐性遗传。随后,我们发现先证者PAX6基因第8外显子上存在1个对蛋白结构和功能可能有显著影响的移码突变c.415dupA(p.R139fs),该变异位点尚未见报道。PAX6基因是先天性虹膜缺损的致病基因,多为常染色体显性遗传,该家系符合家系共分离,杂合子患病和系谱连续传递方式证实此病的常染色体显性遗传方式。此外,先证者父母未携带该变异,也无任何先天性虹膜缺损合并先天性白内障的临床表型,提示这一变异是由于先证者父母在减数分裂中发生错误等原因导致该病的发生,为新发变异,详细机制有待进一步研究。我们通过外显子测序同样检测了与先天性无虹膜有关的ITPR1、ELP4、TRIM44、FOXE3、FOXC1和PITX2基因外显子部分的变异,但未发现可能影响蛋白功能的变异,排除了已报道该病的其他致病基因可能。因此,可以认定该变异位点是导致该家系虹膜缺损合并先天性白内障的原因。此外,本研究中使用基于序列建模的网站swiss-model以及phyre2进行了蛋白质三维结构构建,但因PAX6蛋白仅有36%的同源序列,且均位于N端,后面的序列无法基于同源建模预测。基于目前的模型结果,无法预测该变异影响的蛋白质结构。其他PAX6基因变异导致的先天性虹膜缺损合并先天性白内障家系多表现为完全无虹膜合并白内障[18-21],而该家系患者均存在部分虹膜并伴有眼球震颤。既往PAX6基因移码突变家系多报道为完全无虹膜[22],本研究发现的c.415dupA移码突变还可见周边部分0.2~1.0 mm虹膜。该病患者由于发病年龄小,治疗难度大,生活质量受到较大影响。高通量测序技术在检测罕见遗传病方面发挥着越来越重要的作用,通过高通量测序技术鉴定这些家系中的致病基因及位点能够尽早发现病因,提高优生质量。
本研究发现PAX6基因第8外显子1个新发杂合移码突变c.415dupA(p.R139fs)导致先天性虹膜缺损合并先天性白内障,生物信息学软件预测显示该变异位点位于蛋白质高度保守区,导致mRNA编码错位,使蛋白后续翻译错误,产生异常蛋白而丧失功能。该变异的发现为PAX6基因在虹膜发育中起着不可或缺的作用提供了更多的证据,同时丰富了人类PAX6基因变异谱,为产前诊断和优生优育提供了重要依据。
利益冲突所有作者均声明不存在利益冲突
作者贡献声明顾静、孔艳波:实施研究、收集与分析临床资料、文章撰写;易浩安、江伟阳、杨芳:实施研究、文章撰写;查旭:实施研究、收集与分析临床资料;李凡、何永蜀:参与选题和设计试验、修改文章及定稿
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29 01:57:52
临床输血与检验(2022年3期)2022-06-22 02:52:50
中国典型病例大全(2022年11期)2022-05-13 17:54:50
中国生殖健康(2020年4期)2021-01-18 02:58:10
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
中国生殖健康(2018年4期)2018-11-06 07:12:16
文萃报·周二版(2018年51期)2018-08-04 06:05:18
重庆医学(2015年12期)2015-03-05 05:52:54
